
A Comprehensive Comparative Analysis of Deep Learning Based Feature Representations for Molecular Taste Prediction



Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Preparation
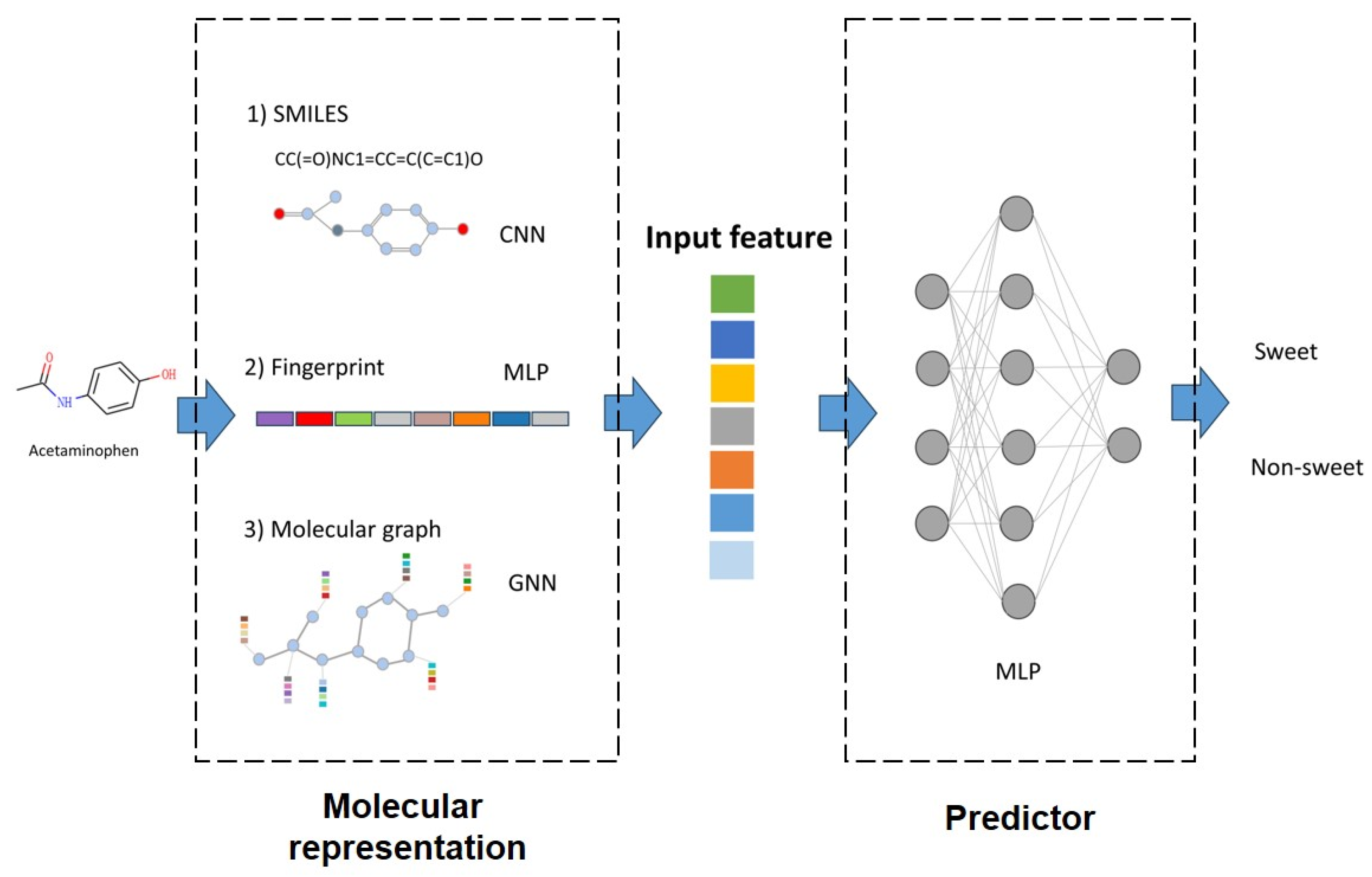
2.2. Molecular Representation
2.2.1. Fingerprint
- (1)
- Morgan fingerprint [28]: A circular fingerprint encoding structural information by considering substructures at different radii around each atom.
- (2)
- PubChem fingerprint [29]: A binary fingerprint derived from the PubChem Compound database, representing molecular structural features based on predefined chemical substructures.
- (3)
- Daylight fingerprint: A descriptor developed by Daylight Chemical Information Systems, encoding chemical features by identifying fragments and substructures within a molecule.
- (4)
- RDKit fingerprint: A fingerprinting method integrated by the RDKit package. It is a dictionary with one entry per bit set in the fingerprint; the keys are the bit IDs; the values are tuples of tuples containing bond indices.
- (5)
- ESPF fingerprint [18]: An explainable substructure partition fingerprint capturing extended connectivity patterns within a molecule, representing the presence of specific atom types and their surrounding environments.
- (6)
- ErG fingerprint [30]: A novel fingerprinting method presented that uses pharmacophore-type node descriptions to encode the relevant molecular properties.
2.2.2. Convolutional Neural Network
- (1)
- Simple CNN [31]:
- (2)
- CNN-LSTM [33]:
- (3)
- CNN-GRU [33]:
2.2.3. Graph Neural Networks
- (1)
- GCN [35]:
- (2)
- NeuralFP [36]:
- (3)
- GIN-AttrMasking [37]:
- (4)
- GIN-ContextPred [37]:
- (5)
- AttentiveFP [38]:
2.3. Predictor
3. Results
3.1. Evaluation Metrics
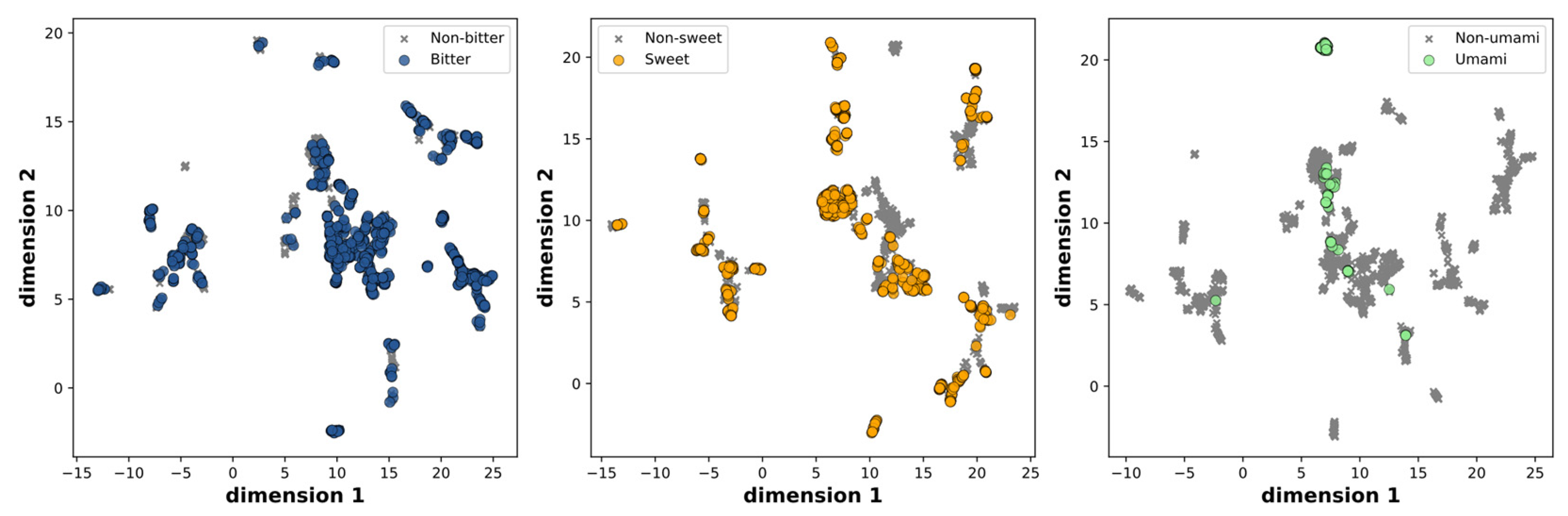
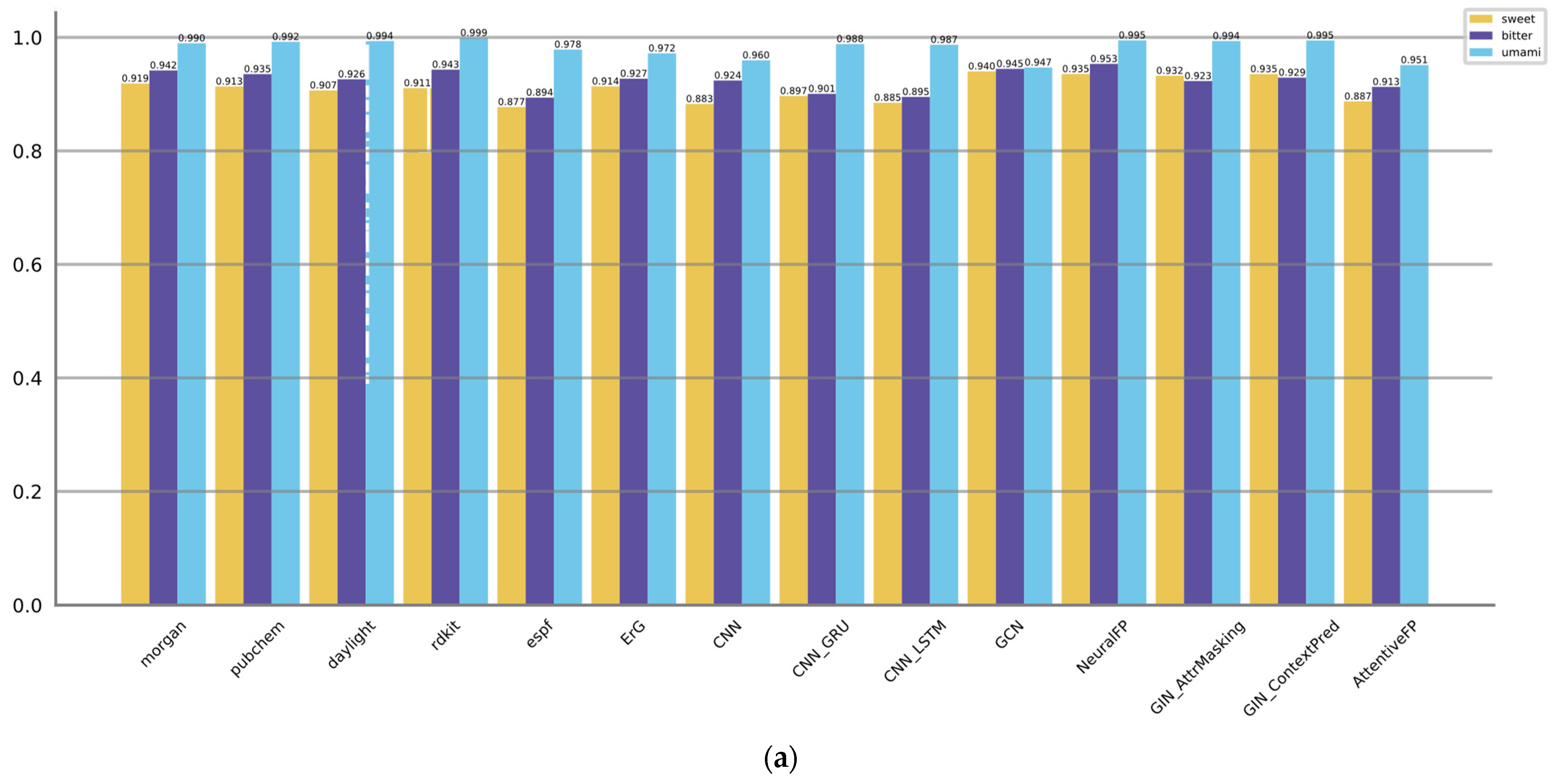
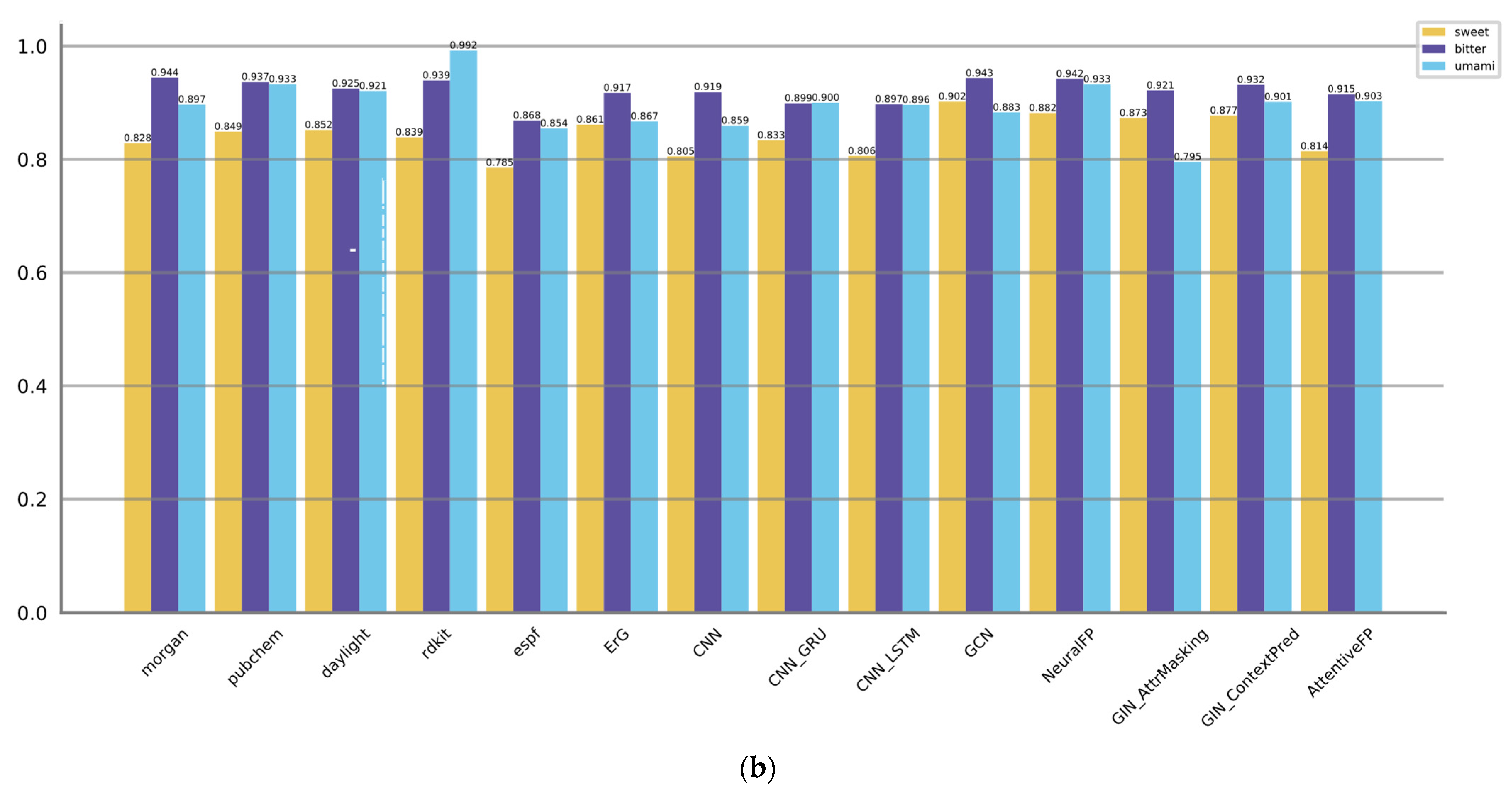
3.2. Comparison of Model Performance
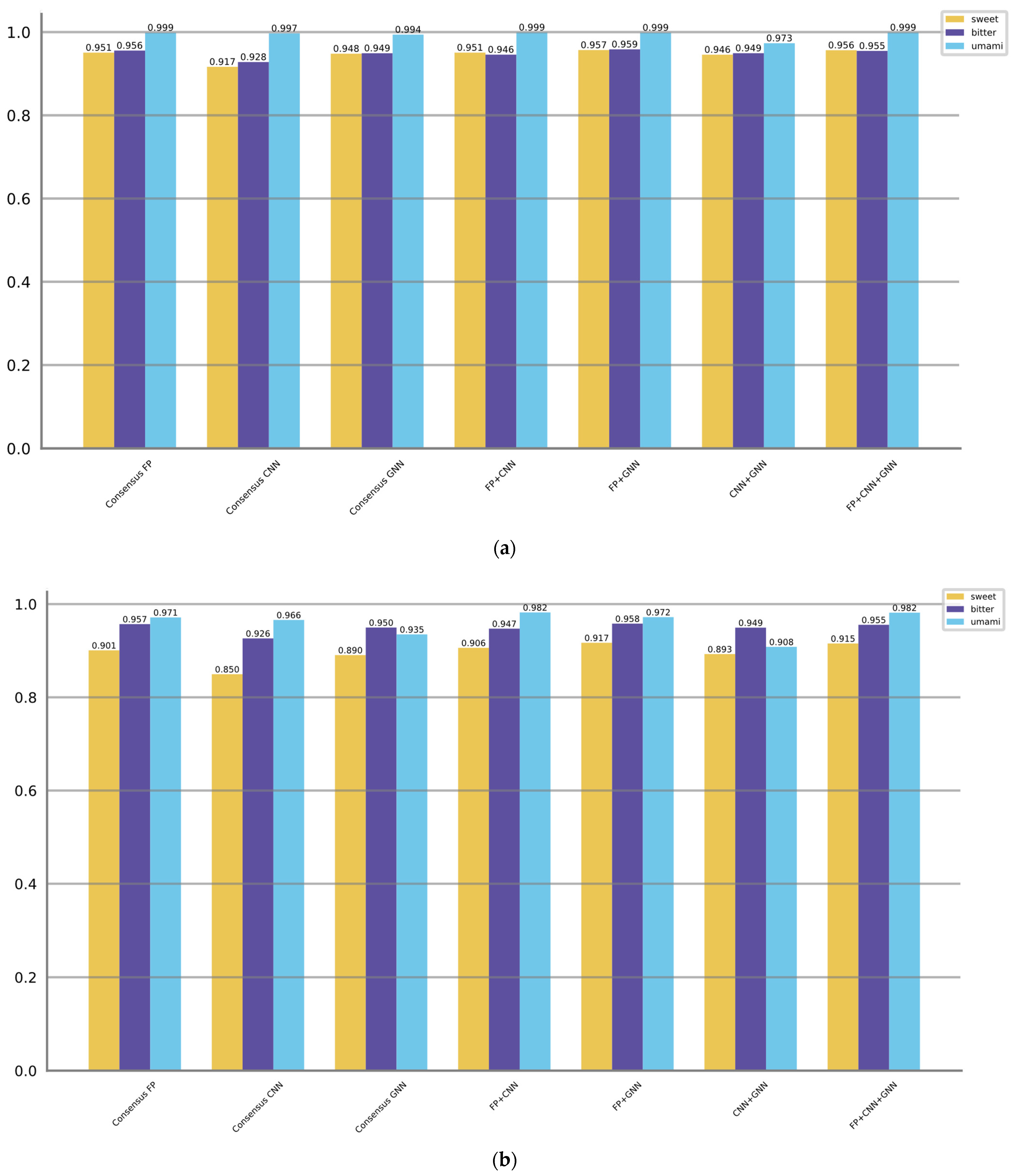
3.3. Voting/Consensus Model Performance
- (1)
- Consensus FP: The ensemble score is obtained by voting from six molecular fingerprint methods.
- (2)
- Consensus CNN: The ensemble score is obtained by voting from three CNN methods.
- (3)
- Consensus GNN: The ensemble score is obtained by voting from five GNN methods.
- (4)
- FP + CNN: This approach combines the top two molecular fingerprint methods and the top two CNN methods based on their best F1 scores.
- (5)
- FP + GNN: This approach combines the top two molecular fingerprint methods and the top two GNN methods based on their best F1 scores.
- (6)
- CNN + GNN: This approach combines the top two CNN methods and the top two GNN methods based on their best F1 scores.
- (7)
- FP + CNN + GNN: This approach combines the top two molecular fingerprint methods, the top two CNN methods, and the top two GNN methods based on their best F1 scores.
3.4. In Silico Compound Taste Database
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chandrashekar, J.; Hoon, M.A.; Ryba, N.J.P.; Zuker, C.S. The Receptors and Cells for Mammalian Taste. Nature 2006, 444, 288–294. [Google Scholar] [CrossRef]
- Drewnowski, A.; Gomez-Carneros, C. Bitter Taste, Phytonutrients, and the Consumer: A Review. Am. J. Clin. Nutr. 2000, 72, 1424–1435. [Google Scholar] [CrossRef]
- Johnson, R.J.; Segal, M.S.; Sautin, Y.; Nakagawa, T.; Feig, D.I.; Kang, D.-H.; Gersch, M.S.; Benner, S.; Sánchez-Lozada, L.G. Potential Role of Sugar (Fructose) in the Epidemic of Hypertension, Obesity and the Metabolic Syndrome, Diabetes, Kidney Disease, and Cardiovascular Disease. Am. J. Clin. Nutr. 2007, 86, 899–906. [Google Scholar] [CrossRef]
- Rojas, C.; Ballabio, D.; Pacheco Sarmiento, K.; Pacheco Jaramillo, E.; Mendoza, M.; García, F. ChemTastesDB: A Curated Database of Molecular Tastants. Food Chem. Mol. Sci. 2022, 4, 100090. [Google Scholar] [CrossRef]
- Banerjee, P.; Preissner, R. BitterSweetForest: A Random Forest Based Binary Classifier to Predict Bitterness and Sweetness of Chemical Compounds. Front. Chem. 2018, 6, 93. [Google Scholar] [CrossRef]
- Goel, M.; Sharma, A.; Chilwal, A.S.; Kumari, S.; Kumar, A.; Bagler, G. Machine Learning Models to Predict Sweetness of Molecules. Comput. Biol. Med. 2023, 152, 106441. [Google Scholar] [CrossRef]
- Fritz, F.; Preissner, R.; Banerjee, P. VirtualTaste: A Web Server for the Prediction of Organoleptic Properties of Chemical Compounds. Nucleic Acids Res. 2021, 49, W679–W684. [Google Scholar] [CrossRef]
- Zheng, S.; Chang, W.; Xu, W.; Xu, Y.; Lin, F. E-Sweet: A Machine-Learning Based Platform for the Prediction of Sweetener and Its Relative Sweetness. Front. Chem. 2019, 7, 35. [Google Scholar] [CrossRef]
- Rojas, C.; Todeschini, R.; Ballabio, D.; Mauri, A.; Consonni, V.; Tripaldi, P.; Grisoni, F. A QSTR-Based Expert System to Predict Sweetness of Molecules. Front. Chem. 2017, 5, 53. [Google Scholar] [CrossRef]
- Zheng, S.; Jiang, M.; Zhao, C.; Zhu, R.; Hu, Z.; Xu, Y.; Lin, F. E-Bitter: Bitterant Prediction by the Consensus Voting From the Machine-Learning Methods. Front. Chem. 2018, 6, 82. [Google Scholar] [CrossRef]
- Tuwani, R.; Wadhwa, S.; Bagler, G. BitterSweet: Building Machine Learning Models for Predicting the Bitter and Sweet Taste of Small Molecules. Sci. Rep. 2019, 9, 7155. [Google Scholar] [CrossRef]
- Bo, W.; Qin, D.; Zheng, X.; Wang, Y.; Ding, B.; Li, Y.; Liang, G. Prediction of Bitterant and Sweetener Using Structure-Taste Relationship Models Based on an Artificial Neural Network. Food Res. Int. 2022, 153, 110974. [Google Scholar] [CrossRef]
- Dagan-Wiener, A.; Nissim, I.; Ben Abu, N.; Borgonovo, G.; Bassoli, A.; Niv, M.Y. Bitter or Not? BitterPredict, a Tool for Predicting Taste from Chemical Structure. Sci. Rep. 2017, 7, 12074. [Google Scholar] [CrossRef]
- Lee, I.; Keum, J.; Nam, H. DeepConv-DTI: Prediction of Drug-Target Interactions via Deep Learning with Convolution on Protein Sequences. PLoS Comput. Biol. 2019, 15, e1007129. [Google Scholar] [CrossRef]
- Xu, L.; Ru, X.; Song, R. Application of Machine Learning for Drug–Target Interaction Prediction. Front. Genet. 2021, 12, 680117. [Google Scholar] [CrossRef]
- Wen, M.; Zhang, Z.; Niu, S.; Sha, H.; Yang, R.; Yun, Y.; Lu, H. Deep-Learning-Based Drug-Target Interaction Prediction. J. Proteome Res. 2017, 16, 1401–1409. [Google Scholar] [CrossRef]
- Huang, K.; Fu, T.; Glass, L.M.; Zitnik, M.; Xiao, C.; Sun, J. DeepPurpose: A Deep Learning Library for Drug–Target Interaction Prediction. Bioinformatics 2021, 36, 5545–5547. [Google Scholar] [CrossRef]
- Ye, Q.; Zhang, X.; Lin, X. Drug–Target Interaction Prediction via Multiple Classification Strategies. BMC Bioinf. 2022, 22, 461. [Google Scholar] [CrossRef]
- Aldeghi, M.; Coley, C.W. A Graph Representation of Molecular Ensembles for Polymer Property Prediction. Chem. Sci. 2022, 13, 10486–10498. [Google Scholar] [CrossRef]
- Fang, X.; Liu, L.; Lei, J.; He, D.; Zhang, S.; Zhou, J.; Wang, F.; Wu, H.; Wang, H. Geometry-Enhanced Molecular Representation Learning for Property Prediction. Nat. Mach. Intell. 2022, 4, 127–134. [Google Scholar] [CrossRef]
- Chen, D.; Gao, K.; Nguyen, D.D.; Chen, X.; Jiang, Y.; Wei, G.-W.; Pan, F. Algebraic Graph-Assisted Bidirectional Transformers for Molecular Property Prediction. Nat. Commun. 2021, 12, 3521. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Zhang, H.; Zhao, D.; Wu, J.; Wang, L. FP-GNN: A Versatile Deep Learning Architecture for Enhanced Molecular Property Prediction. Brief. Bioinform. 2022, 23, bbac408. [Google Scholar] [CrossRef]
- Yang, Q.; Ji, H.; Lu, H.; Zhang, Z. Prediction of Liquid Chromatographic Retention Time with Graph Neural Networks to Assist in Small Molecule Identification. Anal. Chem. 2021, 93, 2200–2206. [Google Scholar] [CrossRef]
- Rohani, A.; Mamarabadi, M. Free Alignment Classification of Dikarya Fungi Using Some Machine Learning Methods. Neural Comput. Appl. 2019, 31, 6995–7016. [Google Scholar] [CrossRef]
- Cui, T.; El Mekkaoui, K.; Reinvall, J.; Havulinna, A.S.; Marttinen, P.; Kaski, S. Gene–Gene Interaction Detection with Deep Learning. Commun. Biol. 2022, 5, 1238. [Google Scholar] [CrossRef]
- Raghunathan, S.; Priyakumar, U.D. Molecular Representations for Machine Learning Applications in Chemistry. Int. J. Quantum Chem. 2022, 122, e26870. [Google Scholar] [CrossRef]
- Kim, J.; Park, S.; Min, D.; Kim, W. Comprehensive Survey of Recent Drug Discovery Using Deep Learning. Int. J. Mol. Sci. 2021, 22, 9983. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Bryant, S.H. PUG-SOAP and PUG-REST: Web Services for Programmatic Access to Chemical Information in PubChem. Nucleic Acids Res 2015, 43, W605–W611. [Google Scholar] [CrossRef]
- Stiefl, N.; Watson, I.A.; Baumann, K.; Zaliani, A. ErG: 2D Pharmacophore Descriptions for Scaffold Hopping. J. Chem. Inf. Model. 2006, 46, 208–220. [Google Scholar] [CrossRef]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet Classification with Deep Convolutional Neural Networks. Commun. Acm 2017, 60, 84–90. [Google Scholar] [CrossRef]
- Tsubaki, M.; Tomii, K.; Sese, J. Compound–Protein Interaction Prediction with End-to-End Learning of Neural Networks for Graphs and Sequences. Bioinformatics 2019, 35, 309–318. [Google Scholar] [CrossRef]
- Cho, K.; van Merriënboer, B.; Gulcehre, C.; Bahdanau, D.; Bougares, F.; Schwenk, H.; Bengio, Y. Learning Phrase Representations Using RNN Encoder–Decoder for Statistical Machine Translation. In Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP), Doha, Qatar, 25–29 October 2014; Association for Computational Linguistics: Doha, Qatar, 2014; pp. 1724–1734. [Google Scholar]
- Li, M.; Zhou, J.; Hu, J.; Fan, W.; Zhang, Y.; Gu, Y.; Karypis, G. DGL-LifeSci: An Open-Source Toolkit for Deep Learning on Graphs in Life Science. ACS Omega 2021, 6, 27233–27238. [Google Scholar] [CrossRef]
- Jiang, B.; Zhang, Z.; Lin, D.; Tang, J.; Luo, B. Semi-Supervised Learning With Graph Learning-Convolutional Networks. In Proceedings of the 2019 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), Long Beach, CA, USA, 15–20 June 2019; pp. 11305–11312. [Google Scholar]
- Duvenaud, D.; Maclaurin, D.; Aguilera-Iparraguirre, J.; Gómez-Bombarelli, R.; Hirzel, T.; Aspuru-Guzik, A.; Adams, R.P. Convolutional Networks on Graphs for Learning Molecular Fingerprints. In Proceedings of the 28th International Conference on Neural Information Processing Systems—Volume 2, Montreal, QC, Canada, 7–12 December 2015; MIT Press: Cambridge, MA, USA, 2015; pp. 2224–2232. [Google Scholar]
- Hu, W.; Liu, B.; Gomes, J.; Zitnik, M.; Liang, P.; Pande, V.; Leskovec, J. Strategies for Pre-Training Graph Neural Networks. arXiv 2020, arXiv:1905.12265. [Google Scholar]
- Xiong, Z.; Wang, D.; Liu, X.; Zhong, F.; Wan, X.; Li, X.; Li, Z.; Luo, X.; Chen, K.; Jiang, H.; et al. Pushing the Boundaries of Molecular Representation for Drug Discovery with the Graph Attention Mechanism. J. Med. Chem. 2020, 63, 8749–8760. [Google Scholar] [CrossRef] [PubMed]
- Malavolta, M.; Pallante, L.; Mavkov, B.; Stojceski, F.; Grasso, G.; Korfiati, A.; Mavroudi, S.; Kalogeras, A.; Alexakos, C.; Martos, V.; et al. A Survey on Computational Taste Predictors. Eur. Food Res. Technol. 2022, 248, 2215–2235. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.; Ballabio, D.; Consonni, V.; Suárez-Estrella, D.; Todeschini, R. Classification-Based Machine Learning Approaches to Predict the Taste of Molecules: A Review. Food Res. Int. 2023, 171, 113036. [Google Scholar] [CrossRef]
- Stepišnik, T.; Škrlj, B.; Wicker, J.; Kocev, D. A Comprehensive Comparison of Molecular Feature Representations for Use in Predictive Modeling. Comput. Biol. Med. 2021, 130, 104197. [Google Scholar] [CrossRef]
- Yu, J.; Wang, J.; Zhao, H.; Gao, J.; Kang, Y.; Cao, D.; Wang, Z.; Hou, T. Organic Compound Synthetic Accessibility Prediction Based on the Graph Attention Mechanism. J. Chem. Inf. Model. 2022, 62, 2973–2986. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, J.; Cao, Z.; Barati Farimani, A. Molecular Contrastive Learning of Representations via Graph Neural Networks. Nat. Mach. Intell. 2022, 4, 279–287. [Google Scholar] [CrossRef]
- Margulis, E.; Slavutsky, Y.; Lang, T.; Behrens, M.; Benjamini, Y.; Niv, M.Y. BitterMatch: Recommendation Systems for Matching Molecules with Bitter Taste Receptors. J. Cheminf. 2022, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. PLoS Comput. Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Training | Validation | Test | Total | |||
|---|---|---|---|---|---|---|---|
| Number | Positive Rate | Number | Positive Rate | Number | Positive Rate | ||
| Sweet | 637 | 0.350 | 91 | 0.350 | 178 | 0.342 | 906 |
| Nonsweet | 1184 | 169 | 342 | 1695 | |||
| Bitter | 769 | 0.422 | 118 | 0.454 | 239 | 0.460 | 1126 |
| Non-bitter | 1052 | 142 | 281 | 1475 | |||
| Umami | 71 | 0.039 | 8 | 0.031 | 19 | 0.037 | 98 |
| Non-umami | 1750 | 252 | 501 | 2503 | |||
| Model | TP | FP | TN | FN | Acc. | Prec. | Sens. | Spec. | F1 |
|---|---|---|---|---|---|---|---|---|---|
| Morgan | 151 | 57 | 285 | 27 | 0.838 | 0.726 | 0.848 | 0.833 | 0.782 |
| Pubchem | 152 | 61 | 281 | 26 | 0.833 | 0.714 | 0.854 | 0.822 | 0.777 |
| Daylight | 120 | 33 | 309 | 58 | 0.825 | 0.784 | 0.674 | 0.904 | 0.725 |
| RDKit | 127 | 35 | 307 | 51 | 0.835 | 0.784 | 0.713 | 0.898 | 0.747 |
| ESPF | 123 | 49 | 293 | 55 | 0.800 | 0.715 | 0.691 | 0.857 | 0.703 |
| ErG | 140 | 45 | 297 | 38 | 0.840 | 0.757 | 0.787 | 0.868 | 0.771 |
| CNN | 141 | 60 | 282 | 37 | 0.813 | 0.701 | 0.792 | 0.825 | 0.744 |
| CNN_GRU | 134 | 43 | 299 | 44 | 0.833 | 0.757 | 0.753 | 0.874 | 0.755 |
| CNN_LSTM | 114 | 29 | 313 | 64 | 0.821 | 0.797 | 0.640 | 0.915 | 0.710 |
| GCN | 148 | 38 | 304 | 30 | 0.869 | 0.796 | 0.831 | 0.889 | 0.813 |
| NeuralFP | 147 | 37 | 305 | 31 | 0.869 | 0.799 | 0.826 | 0.892 | 0.812 |
| GIN_AttrMasking | 154 | 52 | 290 | 24 | 0.854 | 0.748 | 0.865 | 0.848 | 0.802 |
| GIN_ContextPred | 152 | 52 | 290 | 26 | 0.850 | 0.745 | 0.854 | 0.848 | 0.796 |
| AttentiveFP | 98 | 19 | 323 | 80 | 0.810 | 0.838 | 0.550 | 0.944 | 0.664 |
| Model | TP | FP | TN | FN | Acc. | Prec. | Sens. | Spec. | F1 |
|---|---|---|---|---|---|---|---|---|---|
| Morgan | 197 | 30 | 251 | 42 | 0.862 | 0.868 | 0.824 | 0.893 | 0.845 |
| Pubchem | 202 | 26 | 255 | 37 | 0.879 | 0.886 | 0.845 | 0.907 | 0.865 |
| Daylight | 197 | 43 | 238 | 42 | 0.837 | 0.821 | 0.824 | 0.847 | 0.823 |
| RDKit | 203 | 32 | 249 | 36 | 0.869 | 0.864 | 0.849 | 0.886 | 0.857 |
| ESPF | 196 | 53 | 228 | 43 | 0.815 | 0.787 | 0.820 | 0.811 | 0.803 |
| ErG | 190 | 25 | 256 | 49 | 0.858 | 0.884 | 0.795 | 0.911 | 0.837 |
| CNN | 163 | 16 | 265 | 76 | 0.823 | 0.911 | 0.682 | 0.943 | 0.780 |
| CNN_GRU | 167 | 19 | 262 | 72 | 0.825 | 0.898 | 0.699 | 0.932 | 0.786 |
| CNN_LSTM | 173 | 25 | 256 | 66 | 0.825 | 0.874 | 0.724 | 0.911 | 0.792 |
| GCN | 193 | 27 | 254 | 46 | 0.860 | 0.877 | 0.808 | 0.904 | 0.841 |
| NeuralFP | 207 | 22 | 259 | 32 | 0.896 | 0.904 | 0.866 | 0.922 | 0.885 |
| GIN_AttrMasking | 174 | 23 | 258 | 65 | 0.831 | 0.883 | 0.728 | 0.918 | 0.798 |
| GIN_ContextPred | 169 | 14 | 267 | 70 | 0.838 | 0.923 | 0.707 | 0.950 | 0.801 |
| AttentiveFP | 170 | 19 | 262 | 69 | 0.831 | 0.899 | 0.711 | 0.932 | 0.794 |
| Model | TP | FP | TN | FN | Acc. | Prec. | Sens. | Spec. | F1 |
|---|---|---|---|---|---|---|---|---|---|
| Morgan | 15 | 0 | 501 | 4 | 0.992 | 1.000 | 0.789 | 1.000 | 0.882 |
| Pubchem | 17 | 1 | 500 | 2 | 0.994 | 0.944 | 0.895 | 0.998 | 0.919 |
| Daylight | 16 | 5 | 496 | 3 | 0.985 | 0.762 | 0.842 | 0.990 | 0.800 |
| RDKit | 19 | 14 | 487 | 0 | 0.973 | 0.576 | 1.000 | 0.972 | 0.731 |
| ESPF | 15 | 4 | 497 | 4 | 0.985 | 0.789 | 0.789 | 0.992 | 0.789 |
| ErG | 16 | 1 | 500 | 3 | 0.992 | 0.941 | 0.842 | 0.998 | 0.889 |
| CNN | 13 | 3 | 498 | 6 | 0.983 | 0.813 | 0.684 | 0.994 | 0.743 |
| CNN_GRU | 15 | 0 | 501 | 4 | 0.992 | 1.000 | 0.789 | 1.000 | 0.882 |
| CNN_LSTM | 14 | 1 | 500 | 5 | 0.988 | 0.933 | 0.737 | 0.998 | 0.824 |
| GCN | 15 | 0 | 501 | 4 | 0.992 | 1.000 | 0.789 | 1.000 | 0.882 |
| NeuralFP | 17 | 7 | 494 | 2 | 0.982 | 0.708 | 0.895 | 0.986 | 0.791 |
| GIN_AttrMasking | 16 | 10 | 491 | 3 | 0.975 | 0.615 | 0.842 | 0.980 | 0.711 |
| GIN_ContextPred | 16 | 6 | 495 | 3 | 0.983 | 0.727 | 0.842 | 0.988 | 0.780 |
| AttentiveFP | 19 | 0 | 501 | 3 | 0.994 | 1.000 | 0.842 | 1.000 | 0.914 |
| Model | TP | FP | TN | FN | Acc. | Prec. | Sens. | Spec. | F1 |
|---|---|---|---|---|---|---|---|---|---|
| Consensus FP | 141 | 24 | 318 | 37 | 0.883 | 0.855 | 0.792 | 0.930 | 0.822 |
| Consensus CNN | 138 | 41 | 301 | 40 | 0.844 | 0.771 | 0.775 | 0.880 | 0.773 |
| Consensus GNN | 142 | 23 | 319 | 36 | 0.887 | 0.861 | 0.798 | 0.933 | 0.828 |
| FP + CNN | 156 | 40 | 302 | 22 | 0.881 | 0.796 | 0.876 | 0.883 | 0.834 |
| FP + GNN | 153 | 28 | 314 | 25 | 0.898 | 0.845 | 0.860 | 0.918 | 0.852 |
| CNN + GNN | 141 | 26 | 316 | 37 | 0.879 | 0.844 | 0.792 | 0.924 | 0.817 |
| FP + CNN + GNN | 153 | 29 | 313 | 25 | 0.896 | 0.841 | 0.860 | 0.915 | 0.850 |
| Model | TP | FP | TN | FN | Acc. | Prec. | Sens. | Spec. | F1 |
|---|---|---|---|---|---|---|---|---|---|
| Consensus FP | 205 | 27 | 254 | 34 | 0.883 | 0.884 | 0.858 | 0.904 | 0.870 |
| Consensus CNN | 181 | 19 | 262 | 58 | 0.852 | 0.905 | 0.757 | 0.932 | 0.825 |
| Consensus GNN | 188 | 13 | 268 | 51 | 0.877 | 0.935 | 0.787 | 0.954 | 0.855 |
| FP + CNN | 192 | 16 | 265 | 47 | 0.879 | 0.923 | 0.805 | 0.943 | 0.859 |
| FP + GNN | 202 | 17 | 264 | 37 | 0.896 | 0.922 | 0.845 | 0.940 | 0.882 |
| CNN + GNN | 189 | 13 | 268 | 50 | 0.879 | 0.956 | 0.791 | 0.954 | 0.857 |
| FP + CNN + GNN | 197 | 15 | 266 | 42 | 0.890 | 0.929 | 0.824 | 0.947 | 0.874 |
| Model | TP | FP | TN | FN | Acc. | Prec. | Sens. | Spec. | F1 |
|---|---|---|---|---|---|---|---|---|---|
| Consensus FP | 16 | 1 | 500 | 3 | 0.992 | 0.941 | 0.842 | 0.998 | 0.889 |
| Consensus CNN | 16 | 0 | 501 | 3 | 0.994 | 1.000 | 0.842 | 1.000 | 0.914 |
| Consensus GNN | 17 | 1 | 500 | 2 | 0.994 | 0.944 | 0.895 | 0.998 | 0.919 |
| FP + CNN | 15 | 0 | 501 | 4 | 0.992 | 1.000 | 0.789 | 1.000 | 0.882 |
| FP + GNN | 15 | 0 | 501 | 4 | 0.992 | 1.000 | 0.789 | 1.000 | 0.882 |
| CNN + GNN | 16 | 0 | 501 | 3 | 0.994 | 1.000 | 0.842 | 1.000 | 0.914 |
| FP + CNN + GNN | 15 | 0 | 501 | 4 | 0.992 | 1.000 | 0.789 | 1.000 | 0.882 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.; Chang, S.; Tian, J.; Pan, W.; Feng, L.; Ji, H. A Comprehensive Comparative Analysis of Deep Learning Based Feature Representations for Molecular Taste Prediction. Foods 2023, 12, 3386. https://doi.org/10.3390/foods12183386
Song Y, Chang S, Tian J, Pan W, Feng L, Ji H. A Comprehensive Comparative Analysis of Deep Learning Based Feature Representations for Molecular Taste Prediction. Foods. 2023; 12(18):3386. https://doi.org/10.3390/foods12183386
Chicago/Turabian StyleSong, Yu, Sihao Chang, Jing Tian, Weihua Pan, Lu Feng, and Hongchao Ji. 2023. "A Comprehensive Comparative Analysis of Deep Learning Based Feature Representations for Molecular Taste Prediction" Foods 12, no. 18: 3386. https://doi.org/10.3390/foods12183386
APA StyleSong, Y., Chang, S., Tian, J., Pan, W., Feng, L., & Ji, H. (2023). A Comprehensive Comparative Analysis of Deep Learning Based Feature Representations for Molecular Taste Prediction. Foods, 12(18), 3386. https://doi.org/10.3390/foods12183386