
Simultaneous Screening and Quantification of 479 Pesticides in Green Tea by LC-QTOF-MS


Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Apparatus
2.2. Standard Solution Preparation
2.3. Samples Collection and Preparation
2.4. Establishment of Compound Database
2.5. LC-QTOF-MS Parameters
2.6. Method Validation
3. Results and Discussion
3.1. Establishment of Compound Database
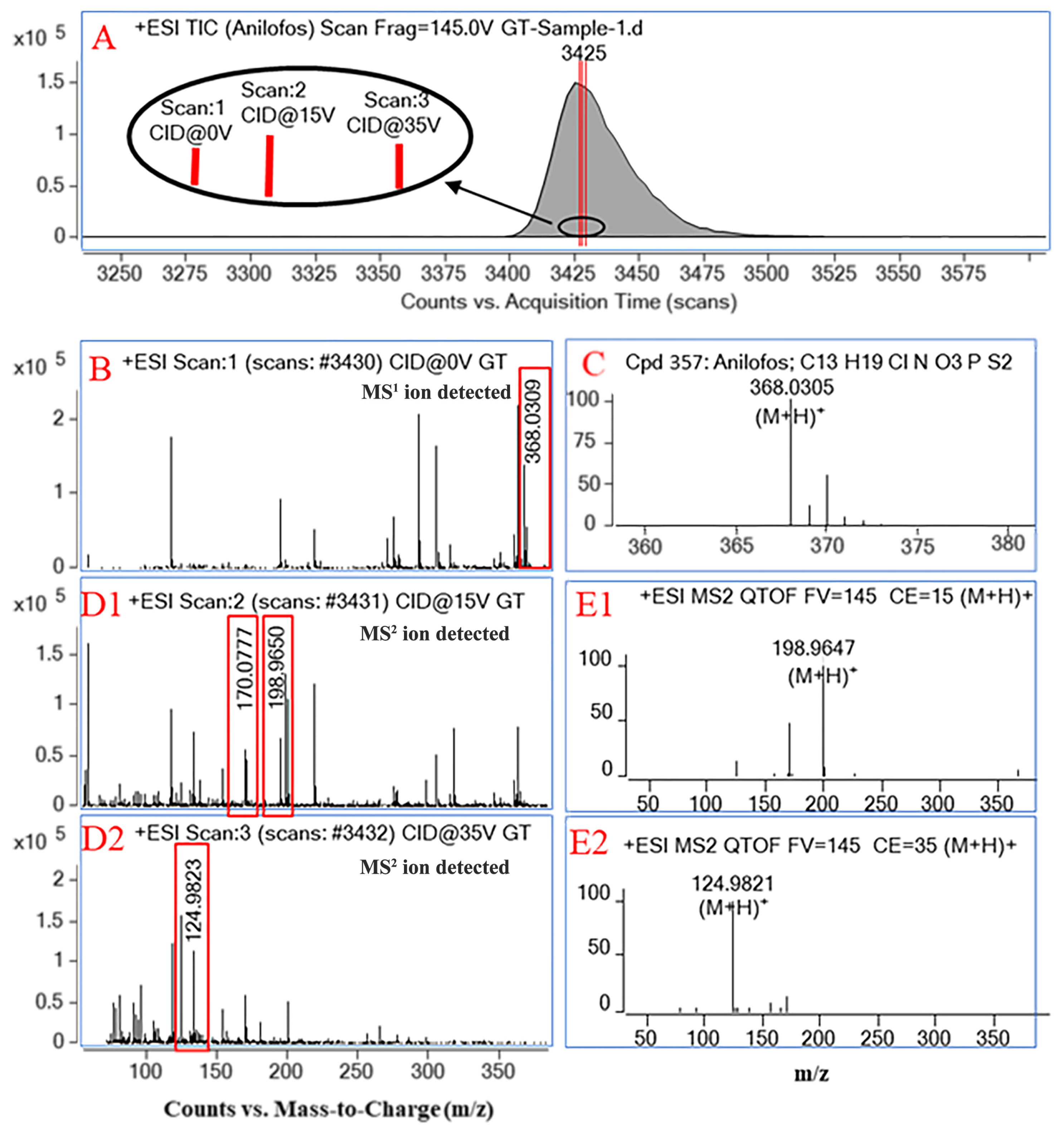
3.2. Development of All Ions MS/MS Method
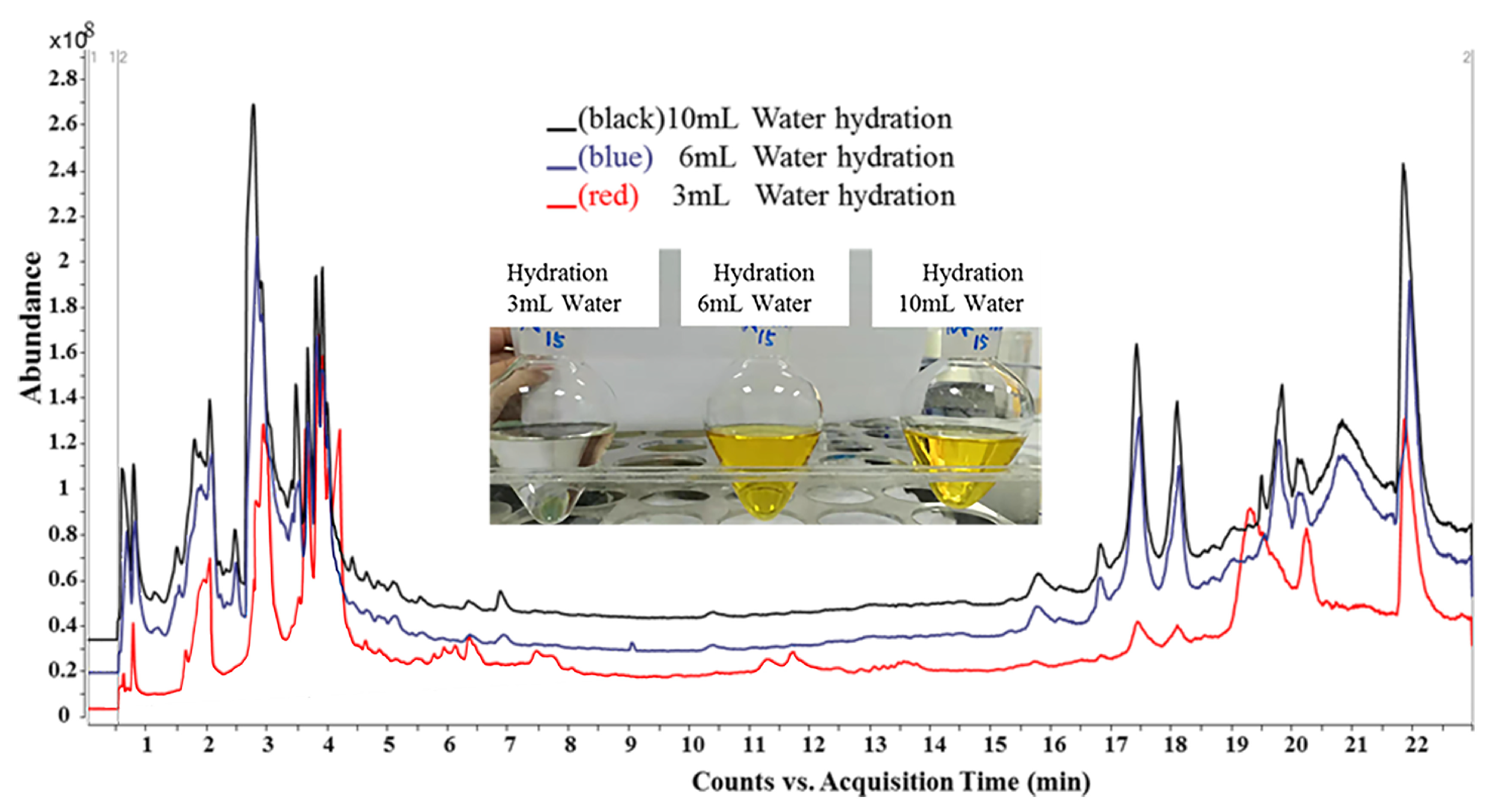
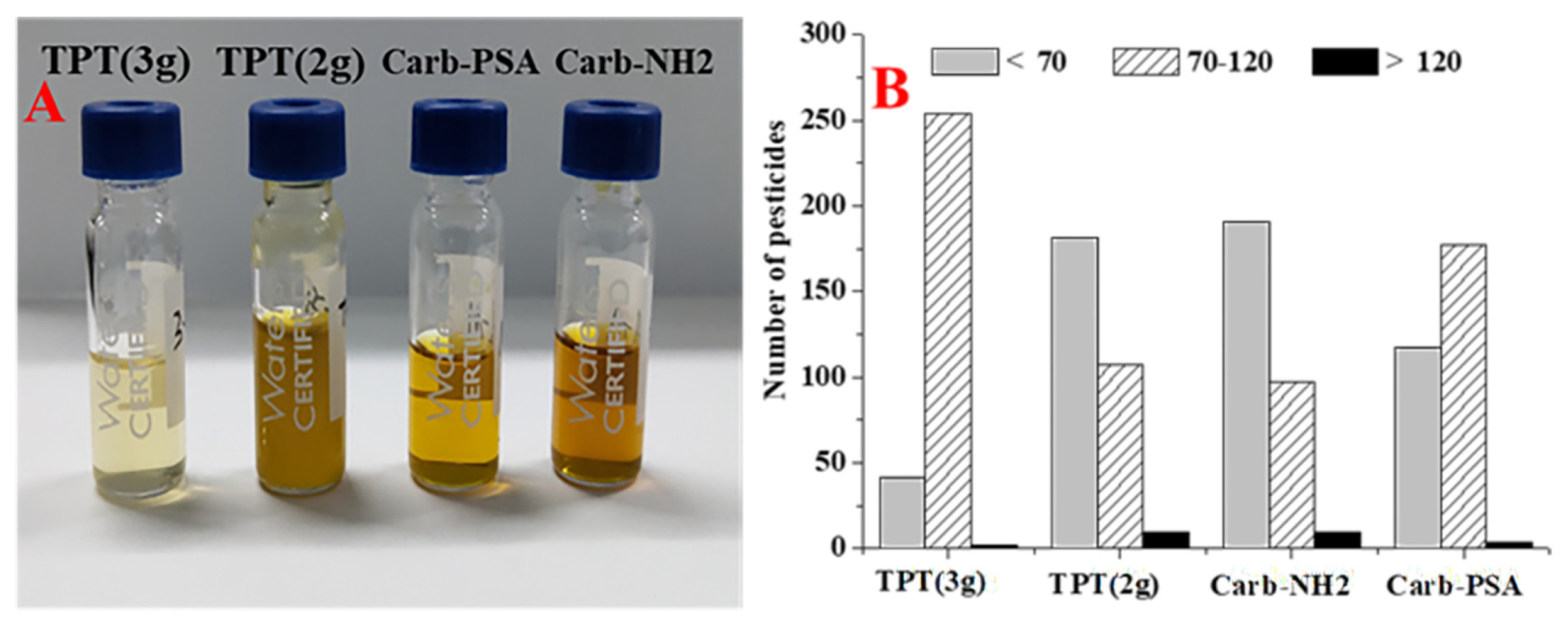
3.3. Optimization in Sample Preparation
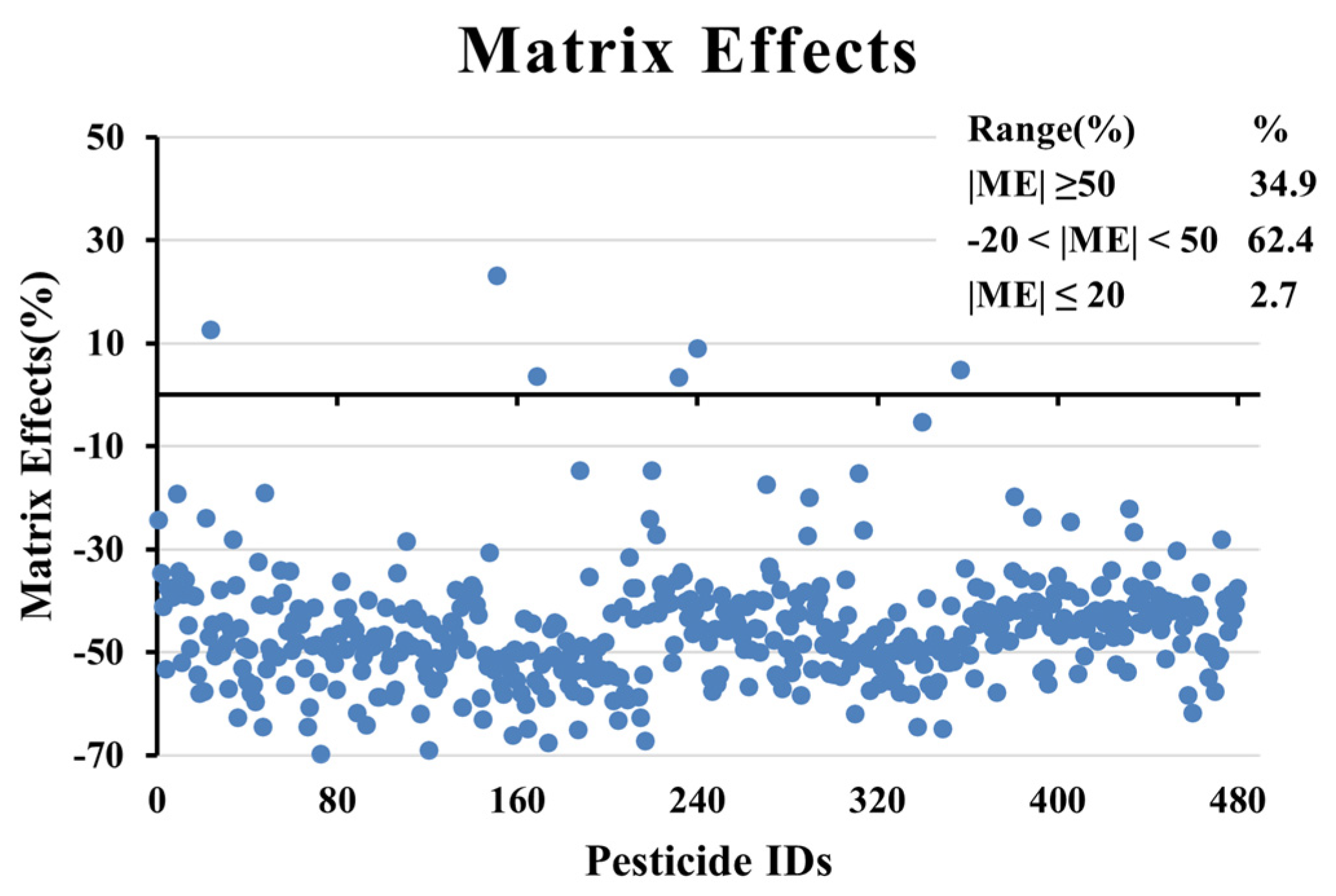
3.4. Matrix Effect
3.5. Method Validation
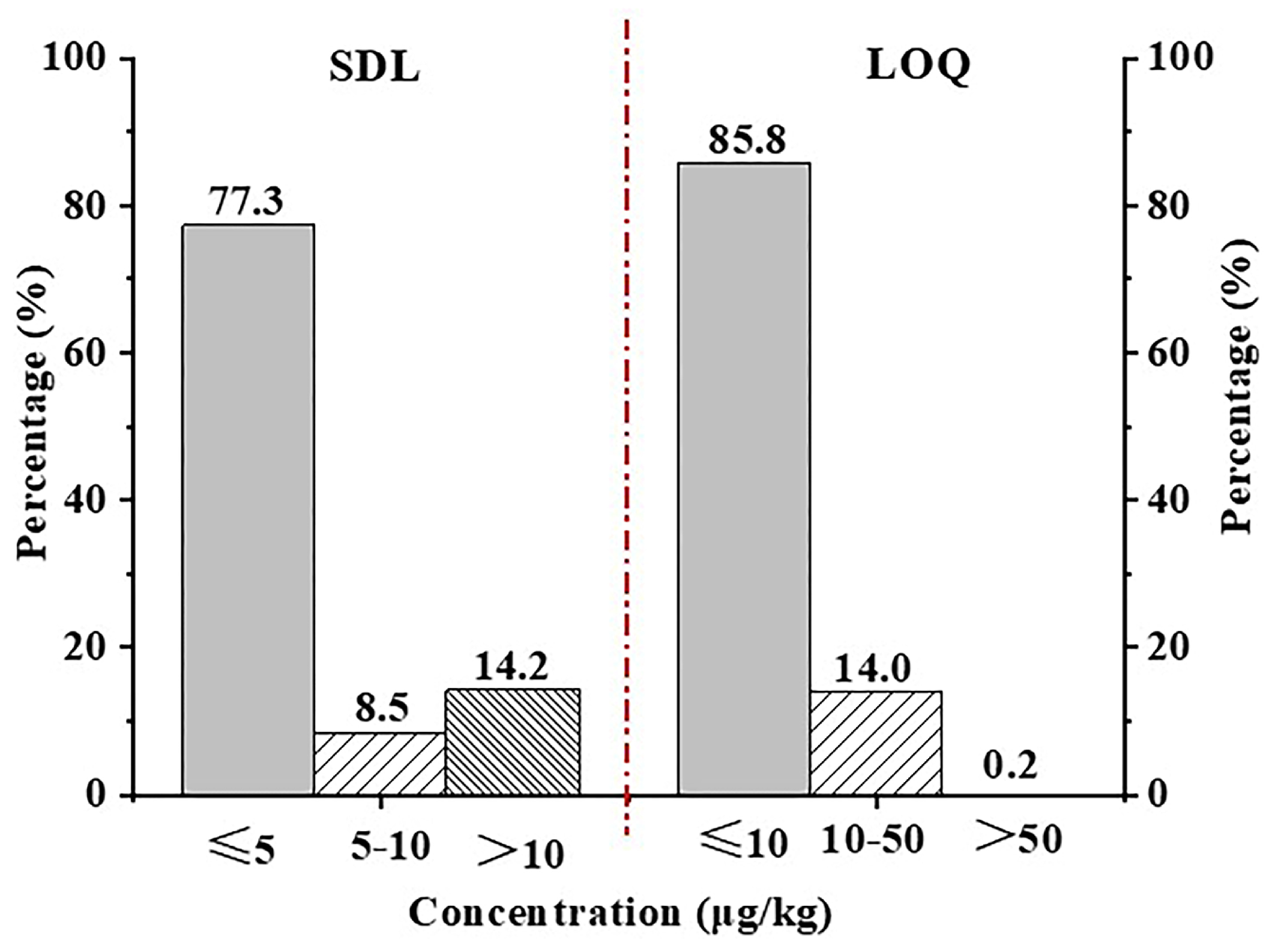
3.5.1. Selectivity, Screening Detection Limit, Limit of Quantification, and Linear Range
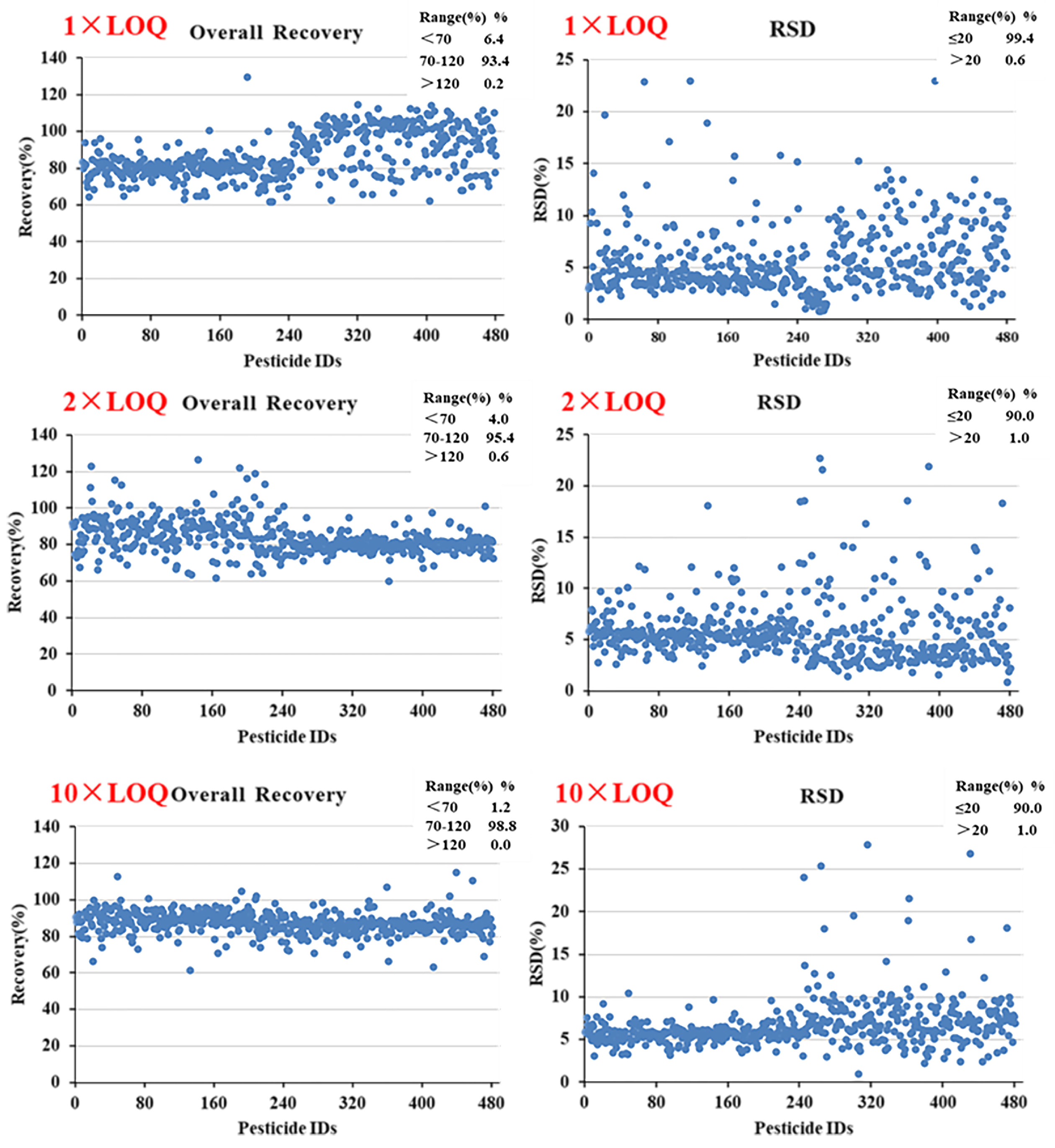
3.5.2. Recovery and Precision
3.5.3. Measurement Uncertainty
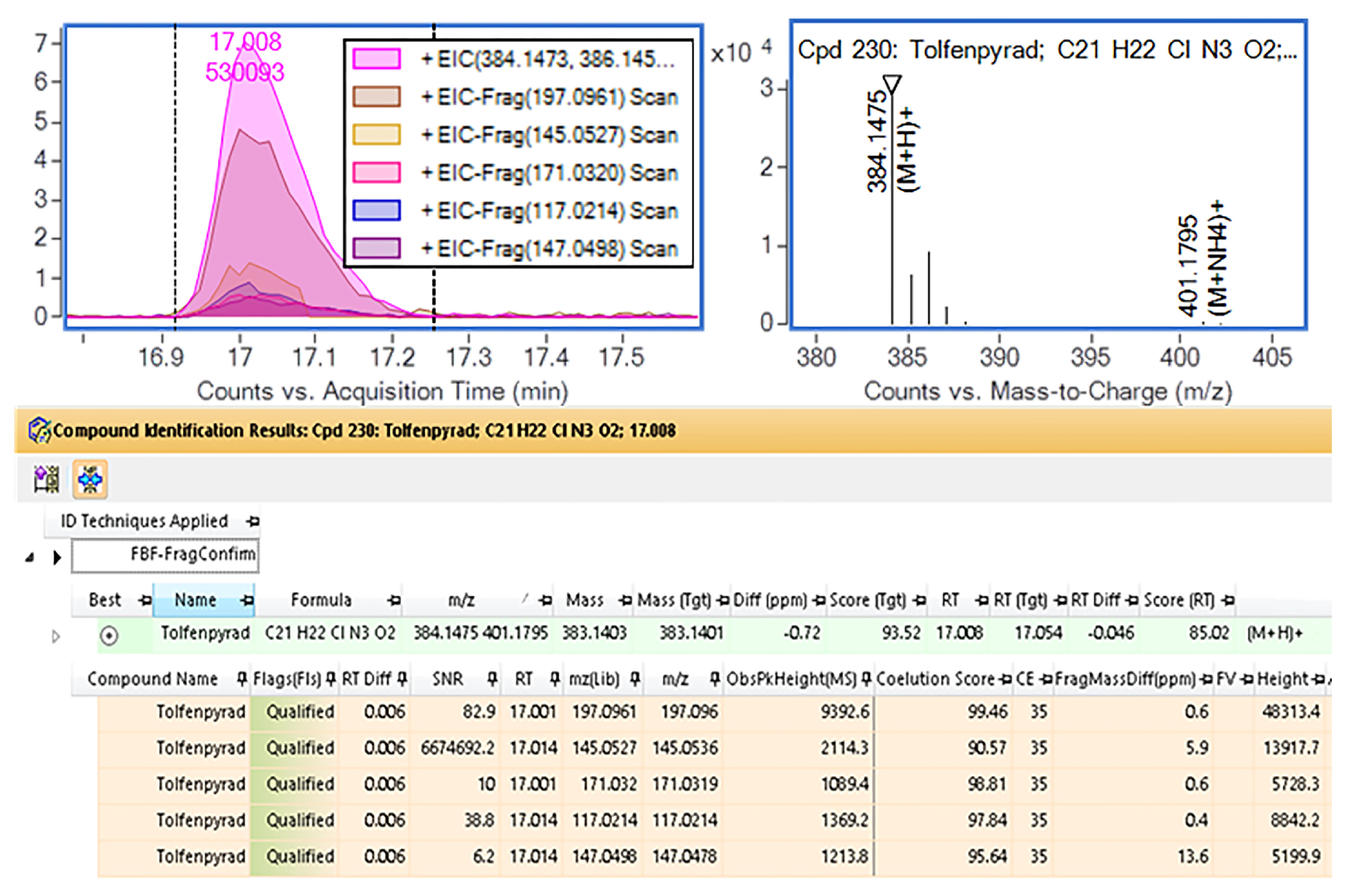
3.6. Analysis of Actual Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malongane, F.; Mcgaw, L.J.; Mudau, F.N. The synergistic potential of various teas, herbs and therapeutic drugs in health improvement: A review. J. Sci. Food Agr. 2017, 97, 4679–4689. [Google Scholar] [CrossRef]
- Dufresne, C.J.; Farnworth, E.R. A review of latest research findings on the health promotion properties of tea. J. Nutr. Biochem. 2001, 12, 404–421. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Fan, Q.; He, X.; Li, W.; Tang, X. Applicability of multifunctional preprocessing device for simultaneous estimation of spreading of green tea, withering of black tea and shaking of oolong tea. J. Sci. Food Agr. 2020, 100, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Food and Agriculture Organization. Food and Agriculture Data. 2020. Available online: http://www.fao.org/faostat/en/#data/QC (accessed on 16 August 2023).
- Chen, H.; Pan, M.; Liu, X.; Lu, C. Evaluation of transfer rates of multiple pesticides from green tea into infusion using water as pressurized liquid extraction solvent and ultra-performance liquid chromatography tandem mass spectrometry. Food Chem. 2017, 216, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Aty, A.M.; Choi, J.H.; Rahman, M.M.; Kim, S.W.; Tosun, A.; Shim, J.H. Residues and contaminants in tea and tea infusions: A review. Food Addit. Contam. A 2014, 31, 1794–1804. [Google Scholar] [CrossRef]
- European Commission. EU Pesticides Database—MRLs. Available online: https://ec.europa.eu/food/plant/pesticides/eu-pesticides-database/start/screen/mrls (accessed on 30 August 2023).
- The Japan Food Chemical Research Foundation. Maximum Residue Limits (MRLs) of Agricultural Chemicals in Foods. Available online: http://db.ffcr.or.jp/front (accessed on 16 August 2023).
- GB 2763-2021; Translation of Maximum Residue Limits for Pesticides in Foods. MOA: Beijing, China, 2021.
- Domínguez, I.; Frenich, A.G.; Romero-González, R. Mass spectrometry approaches to ensure food safety. Anal. Methods 2020, 12, 1148–1162. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, L.; Zhang, X.; Luo, F.; Chen, Z. Transfer of pesticide residue during tea brewing: Understanding the effects of pesticide’s physico-chemical parameters on its transfer behavior. Food Res. Int. 2019, 121, 776–784. [Google Scholar] [CrossRef]
- Cladière, M.; Delaporte, G.; Le Roux, E.; Camel, V. Multi-class analysis for simultaneous determination of pesticides, mycotoxins, process-induced toxicants and packaging contaminants in tea. Food Chem. 2018, 242, 113–121. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Z.; Sun, M.; Zhang, B.; Fan, C. Use of a headspace solid-phase microextraction-based methodology followed by gas chromatography–tandem mass spectrometry for pesticide multiresidue determination in teas. Chromatographia 2018, 81, 809–821. [Google Scholar] [CrossRef]
- Liu, W.; Quan, J. A novel ionic liquid of [BeMIM][Tf2N] for extracting pesticides residues in tea sample by dispersive liquid–liquid microextraction. Chromatographia 2019, 83, 41–51. [Google Scholar] [CrossRef]
- Chen, H.; Gao, G.; Chai, Y.; Ma, G.; Hao, Z.; Wang, C.; Liu, X.; Lu, C. Multiresidue method for the rapid determination of pesticide residues in tea using ultra performance liquid chromatography Orbitrap high resolution mass spectrometry and in-syringe dispersive solid phase extraction. ACS Omega 2017, 2, 5917–5927. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Tang, H.; Chen, D.; Li, L. A novel method based on MSPD for simultaneous determination of 16 pesticide residues in tea by LC–MS/MS. J. Chromatogr. B 2015, 998, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Zhao, M.; Mao, Q.; Fang, C.; Chen, D.; Yan, P. Rapid one-step cleanup method to minimize matrix effects for residue analysis of alkaline pesticides in tea using liquid chromatography–high resolution mass spectrometry. Food Chem. 2019, 299, 125146. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Teng, X.; Wang, W.; Zhang, Z.; Fan, C. Determination of multiple pesticide residues in teas by gas chromatography with accurate time-of-flight mass spectrometry. J. Sep. Sci. 2019, 42, 1990–2002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chang, Q.; Li, J.; Qiu, G.; Wu, F.; Zhu, R.; Wang, X.; Su, M. A liquid chromatography-time-of-flight/mass spectrometry method for analysis of pesticides and transfer behavior in Radix Codonopsis and Angelica sinensis decoctions. Anal. Methods 2023, 15, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, X.; Liu, P.; Jia, Q.; Han, H.; Jiang, C.; Qiu, J. Determination of three typical metabolites of pyrethroid pesticides in tea using a modified QuEChERS sample preparation by ultra-high performance liquid chromatography tandem mass spectrometry. Foods 2021, 10, 189. [Google Scholar] [CrossRef]
- Pang, G.; Fan, C.; Cao, Y.; Yan, F.; Li, Y.; Kang, J.; Chen, H.; Chang, Q. High throughput analytical techniques for the determination and confirmation of residues of 653 multiclass pesticides and chemical pollutants in tea by GC/MS, GC/MS/MS, and LC/MS/MS: Collaborative study, first action 2014.09. J. AOAC Int. 2015, 98, 1428–1454. [Google Scholar] [CrossRef]
- Chen, Y.; Lopez, S.; Hayward, D.G.; Park, H.Y.; Wong, J.W.; Kim, S.S.; Wan, J.; Reddy, R.M.; Quinn, D.J.; Striniger, D. Determination of multiresidue pesticides in botanical dietary supplements using gas chromatography–triple-quadrupole mass spectrometry (GC-MS/MS). J. Agric. Food Chem. 2016, 64, 6125–6132. [Google Scholar] [CrossRef]
- Ly, T.K.; Ho, T.D.; Behra, P.; Nhu-Trang, T.T. Determination of 400 pesticide residues in green tea leaves by UPLC-MS/MS and GC-MS/MS combined with QuEChERS extraction and mixed-mode SPE clean-up method. Food Chem. 2020, 326, 126928. [Google Scholar] [CrossRef]
- Chen, G.; Cao, P.; Liu, R. A multi-residue method for fast determination of pesticides in tea by ultra performance liquid chromatography-electrospray tandem mass spectrometry combined with modified QuEChERS sample preparation proce-dure. Food Chem. 2011, 125, 1406411. [Google Scholar] [CrossRef]
- Hayward, D.G.; Wong, J.W.; Shi, F.; Zhang, K.; Lee, N.S.; DiBenedetto, A.L.; Hengel, M.J. Multiresidue pesticide analysis of botanical dietary supplements using salt-out acetonitrile extraction, solid-phase extraction cleanup column, and gas chromatography-triple quadrupole mass spectrometry. Anal. Chem. 2013, 85, 4686–4693. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Lei, S.; Guo, L.; Qiu, S. Optimization of a multi-residue method for 101 pesticides in green tea leaves using gas chromatography-tandem mass spectrometry. Braz. J. Pharm. Sci. 2016, 26, 401–407. [Google Scholar] [CrossRef]
- Pang, G.F.; Fan, C.L.; Chang, Q.Y.; Li, J.X.; Kang, J. Screening of 485 pesticide residues in fruits and vegetables by liquid chromatography-quadrupole-time-of-flight mass spectrometry based on TOF accurate mass database and QTOF spectrum library. J. AOAC Int. 2018, 101, 1156–1182. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Zhang, Y.; Zhou, Y.; Li, G.; Feng, X. Technical overview of orbitrap high resolution mass spectrometry and its application to the detection of small molecules in food (update since 2012). Crit. Rev. Anal. Chem. 2020, 52, 1–34. [Google Scholar] [CrossRef]
- Yu, L.; Song, W.; Lu, Y.; Zhao, M.; Zhou, F.; Hu, Y.; Zheng, P. Rapid determination of 204 pesticide residues in tea by ultra performance liquid chromatography coupled with quadrupole-time of flight mass spectrometry. Chin. J. Chromatogr. 2015, 33, 597–612. [Google Scholar] [CrossRef]
- Martínez-Domínguez, G.; Romero-González, R.; Frenich, A.G. Multi-class methodology to determine pesticides and mycotoxins in green tea and royal jelly supplements by liquid chromatography coupled to Orbitrap high resolution mass spectrometry. Food Chem. 2016, 197, 907–915. [Google Scholar] [CrossRef]
- Saito-Shida, S.; Hamasaka, T.; Nemoto, S.; Akiyama, H. Multiresidue determination of pesticides in tea by liquid chromatography-high-resolution mass spectrometry: Comparison between Orbitrap and time-of-flight mass analyzers. Food Chem. 2018, 256, 140–148. [Google Scholar] [CrossRef]
- Xie, G.R.; Huang, J.T.; Sung, G.; Chang, J.; Chen, H.J. Traceable and Integrated Pesticide Screening (TIPS), a Systematic and Retrospective Strategy for Screening 900 Pesticides and Unknown Metabolites in Tea. Anal. Chem. 2022, 94, 16647–16657. [Google Scholar] [CrossRef]
- European Commission SANTE 11312/2021; Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed. European Commission: Brussels, Belgium, 2021; pp. 1–57. Available online: https://www.eurl-pesticides.eu/userfiles/file/EurlALL/SANTE_11312_2021.pdf (accessed on 16 August 2023).
- de Oliveira Arias, J.L.; Rombaldi, C.; Caldas, S.S.; Primel, E.G. Alternative sorbents for the dispersive solid-phase extraction step in quick, easy, cheap, effective, rugged and safe method for extraction of pesticides from rice paddy soils with determination by liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2014, 1360, 66–75. [Google Scholar] [CrossRef]
- Eyken, V.A.; Furlong, D.; Arooni, S.; Butterworth, F.; Roy, J.F.; Zweigenbaum, J.; Bayen, S. Direct injection high performance liquid chromatography coupled to data independent acquisition mass spectrometry for the screening of antibiotics in honey. J. Food Drug Anal. 2019, 27, 679–691. [Google Scholar] [CrossRef]
- Wang, F.; Li, S.; Feng, H.; Yang, Y.; Xiao, B.; Chen, D. An enhanced sensitivity and cleanup strategy for the nontargeted screening and targeted determination of pesticides in tea using modified dispersive solid-phase extraction and cold-induced acetonitrile aqueous two-phase systems coupled with liquid chromatography-high resolution mass spectrometry. Food Chem. 2019, 275, 530–538. [Google Scholar] [CrossRef]
- Stahnke, H.; Kittlaus, S.; Kempe, G.; Alderet, L. Reduction of matrix effects in liquid chromatography–electrospray ionization–mass spectrometry by dilution of the sample extracts: How much dilution is needed? Anal. Chem. 2012, 84, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Uclés, S.; Lozano, A.; Sosa, A.; Vázquez, P.P.; Valverde, A.; Fernández-Alba, A.R. Matrix interference evaluation employing GC and LC coupled to triple quadrupole tandem mass spectrometry. Talanta 2017, 174, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Valverde, A.; Aguilera, A.; Valverde-Monterreal, A. Practical and valid guidelines for realistic estimation of measurement uncertainty in multi-residue analysis of pesticides. Food Control 2017, 71, 1–9. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pesticide | Concentration Range (mg/kg) | LOQ (mg/kg) | EU-MRL (mg/kg) | China-MRL (mg/kg) | Exceeding MRL Samples | |
|---|---|---|---|---|---|---|
| Number | % | |||||
| Buprofezin | 0.138–0.529 | 0.005 | 0.05 | 10 | 5 | 5.3 |
| Pyridaben | 0.125 | 0.005 | 0.05 | 5 | 1 | 1.1 |
| Abamectin | 0.262 | 0.005 | 0.05 | / | 1 | 1.1 |
| Chlorpyrifos | 0.046–0.731 | 0.005 | 0.01 | 2 | 7 | 7.4 |
| Triadimenol | 0.223–1.400 | 0.005 | 0.05 | / | 8 | 8.4 |
| Propiconazole | 0.064 | 0.005 | 0.05 | / | 1 | 1.1 |
| Chlorantraniliprole | 0.106 | 0.005 | 0.02 | / | 1 | 1.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Xie, Y.; Tong, K.; Chang, Q.; Hu, X.; Fan, C.; Chen, H. Simultaneous Screening and Quantification of 479 Pesticides in Green Tea by LC-QTOF-MS. Foods 2023, 12, 4177. https://doi.org/10.3390/foods12224177
Wu X, Xie Y, Tong K, Chang Q, Hu X, Fan C, Chen H. Simultaneous Screening and Quantification of 479 Pesticides in Green Tea by LC-QTOF-MS. Foods. 2023; 12(22):4177. https://doi.org/10.3390/foods12224177
Chicago/Turabian StyleWu, Xingqiang, Yujie Xie, Kaixuan Tong, Qiaoying Chang, Xueyan Hu, Chunlin Fan, and Hui Chen. 2023. "Simultaneous Screening and Quantification of 479 Pesticides in Green Tea by LC-QTOF-MS" Foods 12, no. 22: 4177. https://doi.org/10.3390/foods12224177
APA StyleWu, X., Xie, Y., Tong, K., Chang, Q., Hu, X., Fan, C., & Chen, H. (2023). Simultaneous Screening and Quantification of 479 Pesticides in Green Tea by LC-QTOF-MS. Foods, 12(22), 4177. https://doi.org/10.3390/foods12224177