
Insights into the Protein Differentiation Mechanism between Jinhua Fatty Ham and Lean Ham through Label-Free Proteomics

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Processing of Fatty Ham and Lean Ham
2.2. Protein Extraction and Digestion
2.3. HPLC-MS/MS Analysis
2.4. Protein Identification and Quantitative Analysis
2.5. Bioinformatic Analysis
2.6. Statistical Analysis
3. Results
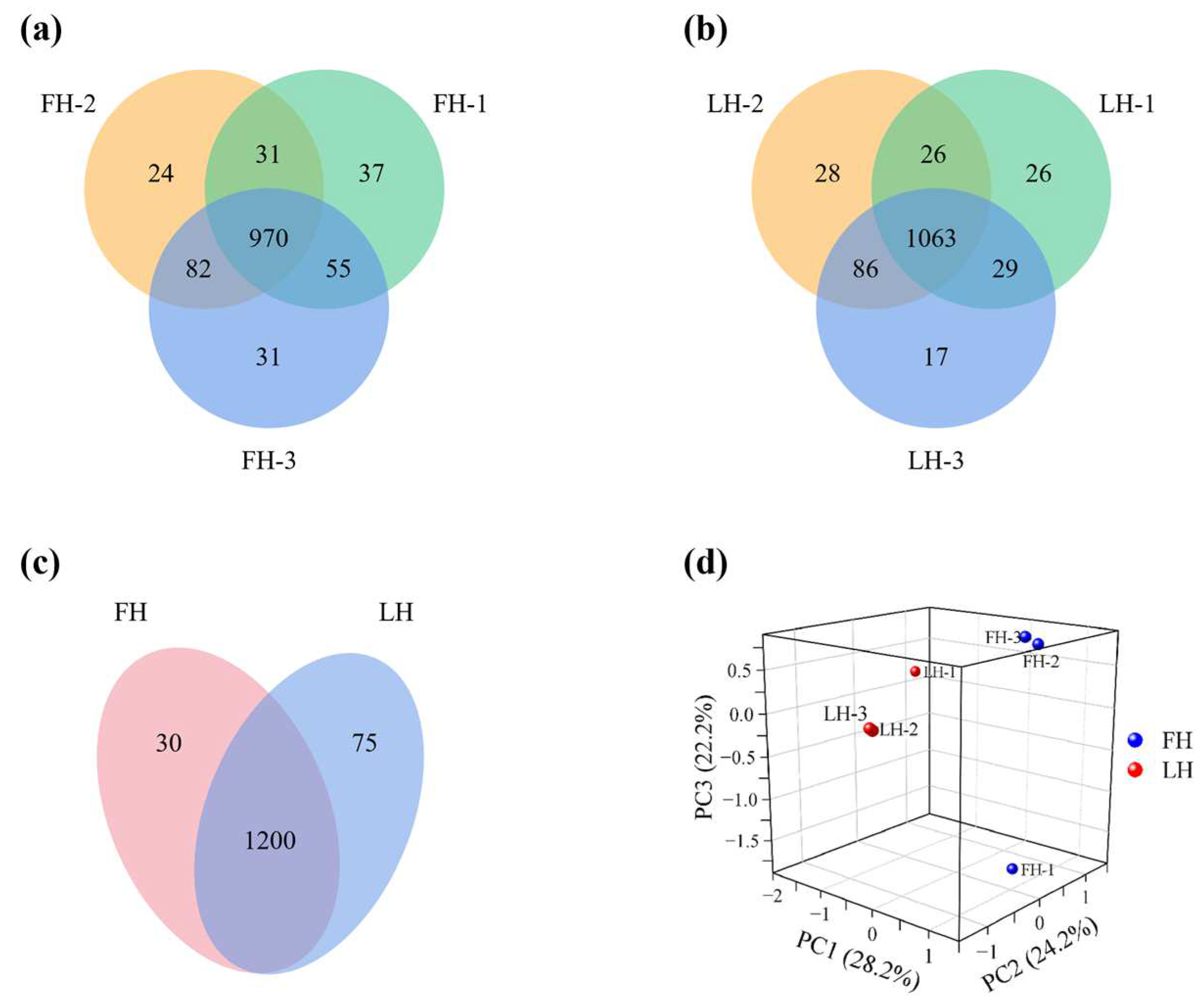
3.1. Identification of Differential Proteins (DPs)
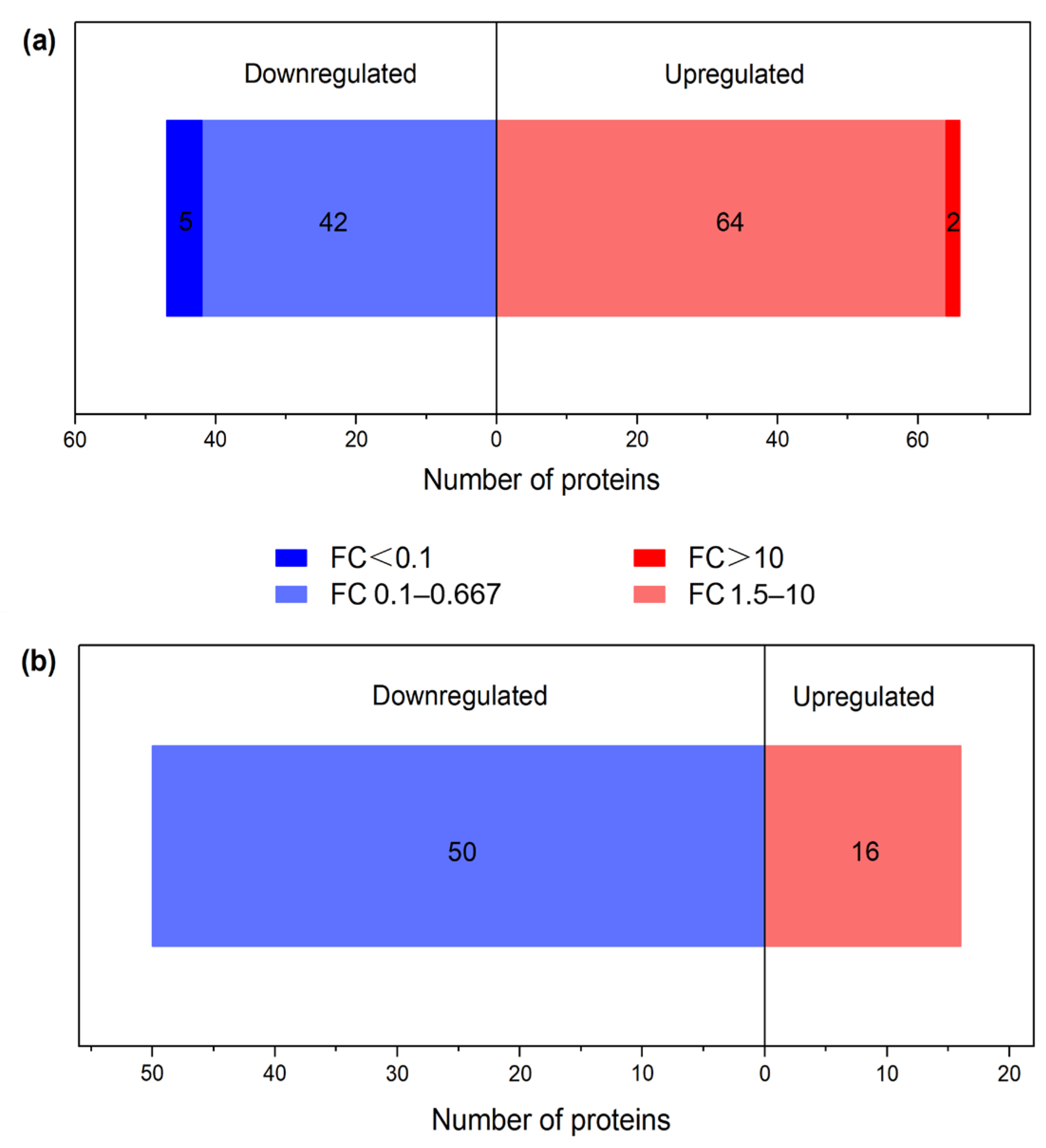
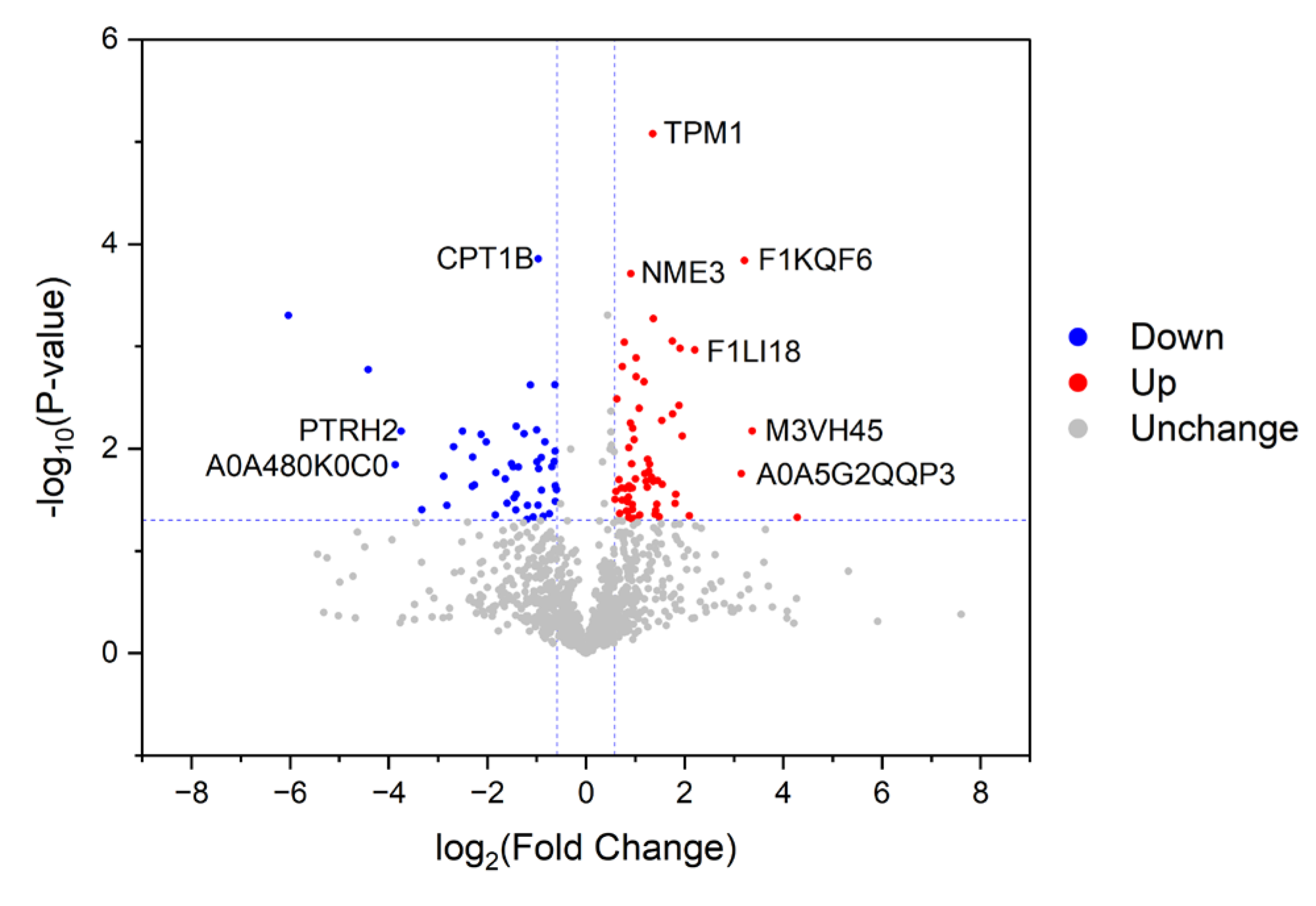
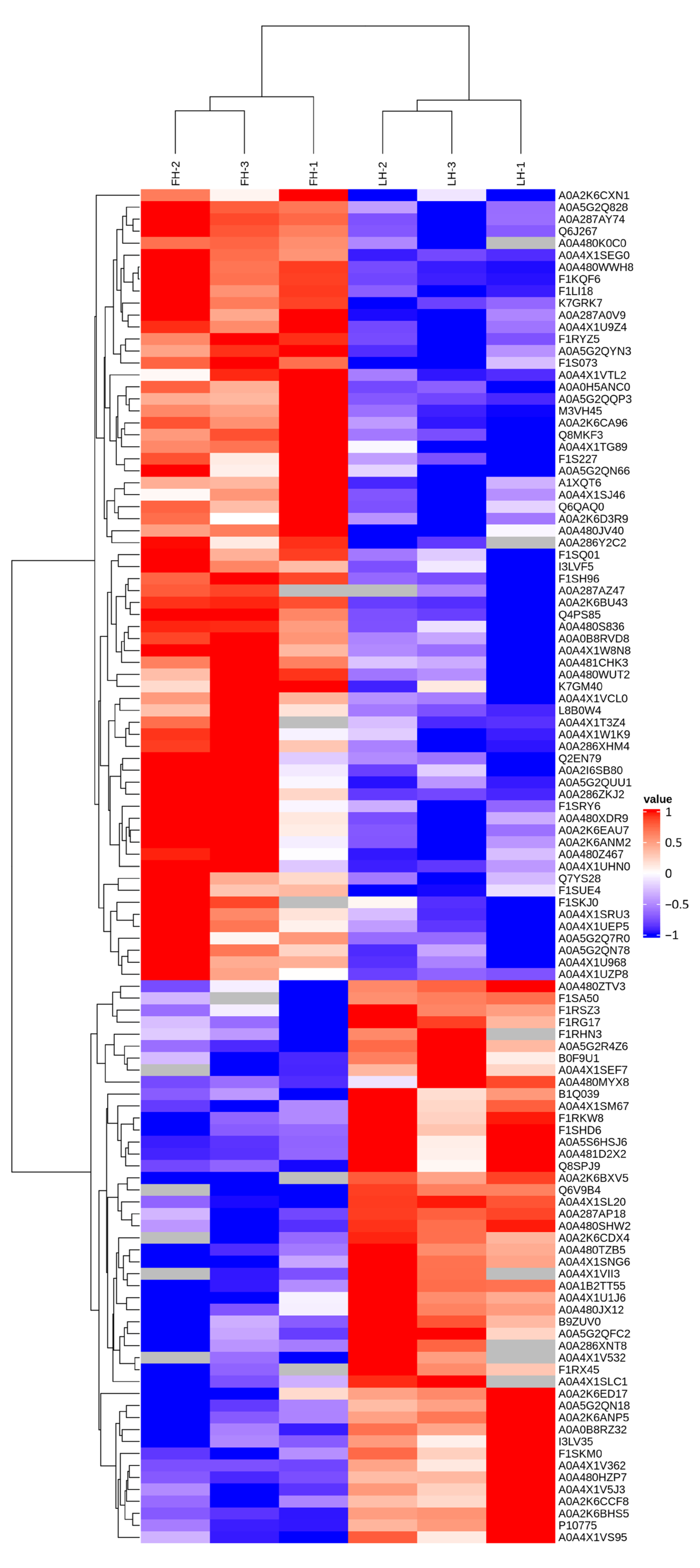
3.2. Characterization of Proteins with Differential Abundances
3.3. Subcellular Localization
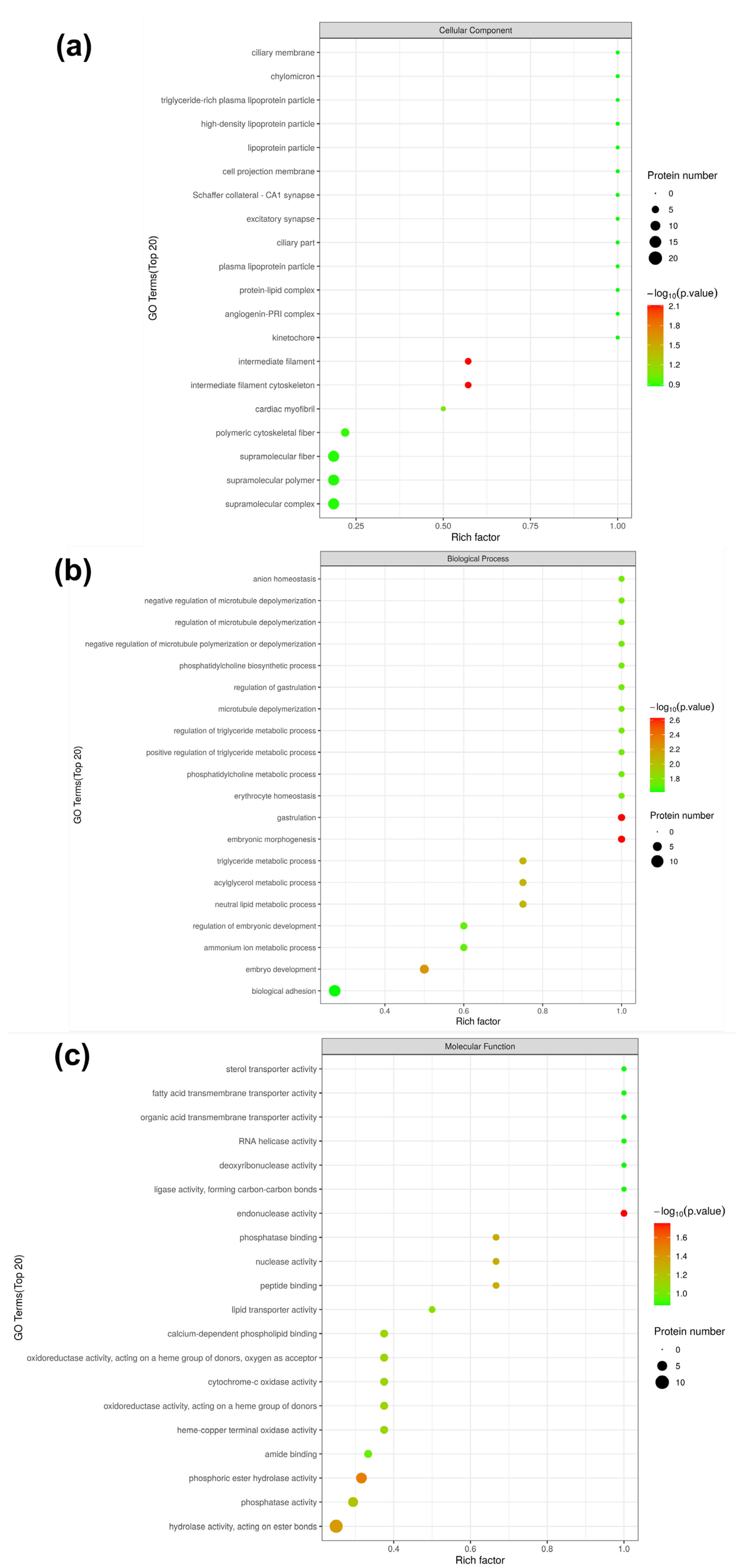
3.4. Gene Ontology (GO) Analysis
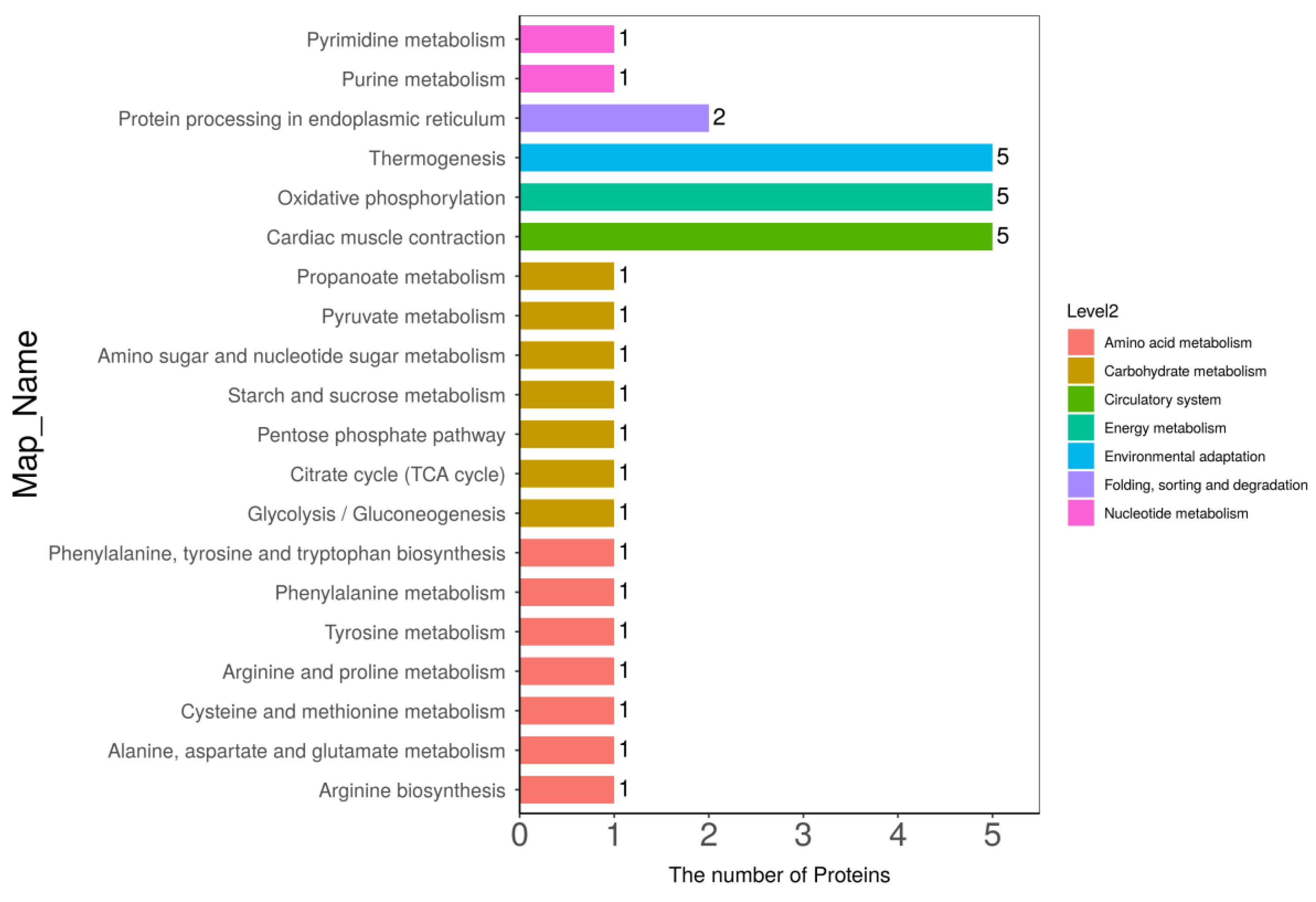
3.5. Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analysis
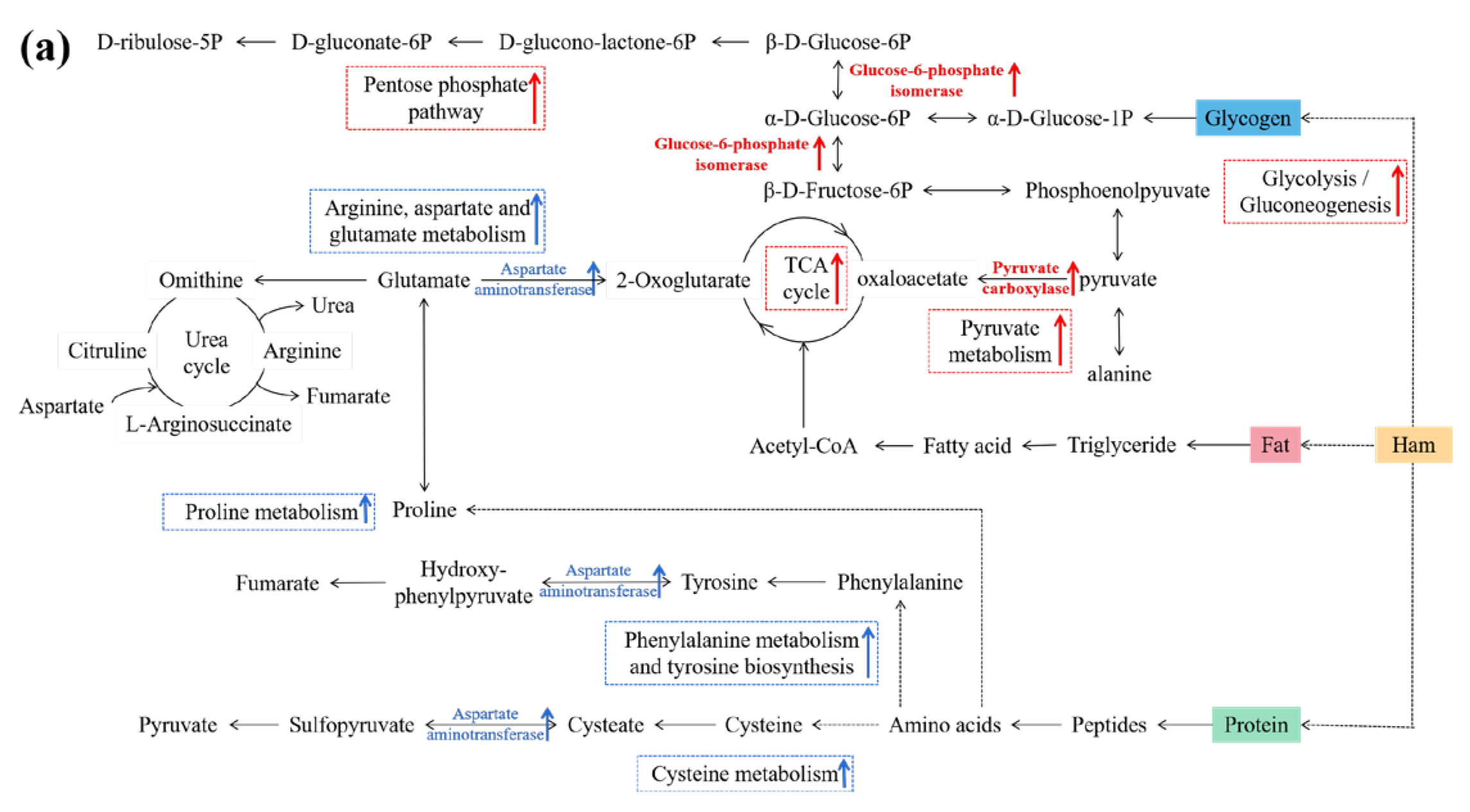
3.6. Connections between DPs and Metabolic Pathways
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Li, F.; Chen, J.; Sun, Z.; Wang, F.; Wang, C.; Fu, L. High-throughput sequencing-based characterization of the predominant microbial community associated with characteristic flavor formation in Jinhua Ham. Food Microbiol. 2021, 94, 103643. [Google Scholar] [CrossRef]
- Martinez-Onandi, N.; Rivas-Canedo, A.; Avila, M.; Garde, S.; Nunez, M.; Picon, A. Influence of physicochemical characteristics and high pressure processing on the volatile fraction of Iberian dry-cured ham. Meat Sci. 2017, 131, 40–47. [Google Scholar] [CrossRef]
- Lee, H.C.; Wonderly, M.P.; Pokharel, S.; Strasburg, G.M.; Marks, B.P.; Fernando, R.H.; Kang, I. Cold-batter mincing of hot-boned and crust-freeze-air-chilled ham muscle reduced fat content in protein gels. Int. J. Food Sci. Technol. 2020, 55, 3267–3277. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, K.; Li, H.; Li, S.; Xu, W.; Chen, L.; Xie, J.; Tang, H. Physicochemical property, volatile flavor quality, and microbial community composition of Jinhua fatty ham and lean ham: A comparative study. Front. Microbiol. 2023, 14, 1124770. [Google Scholar] [CrossRef]
- Zhou, C.Y.; Wang, C.; Tang, C.B.; Dai, C.; Bai, Y.; Yu, X.B.; Li, C.B.; Xu, X.L.; Zhou, G.H.; Cao, J.X. Label-free proteomics reveals the mechanism of bitterness and adhesiveness in Jinhua ham. Food Chem. 2019, 297, 125012. [Google Scholar] [CrossRef]
- Lopez-Pedrouso, M.; Perez-Santaescolastica, C.; Franco, D.; Fulladosa, E.; Carballo, J.; Zapata, C.; Lorenzo, J.M. Comparative proteomic profiling of myofibrillar proteins in dry-cured ham with different proteolysis indices and adhesiveness. Food Chem. 2018, 244, 238–245. [Google Scholar] [CrossRef]
- Gallego, M.; Mora, L.; Aristoy, M.C.; Toldra, F. Optimisation of a simple and reliable label-free methodology for the relative quantitation of raw pork meat proteins. Food Chem. 2015, 182, 74–80. [Google Scholar] [CrossRef]
- Zhou, C.-Y.; Wang, C.; Dai, C.; Bai, Y.; Yu, X.-B.; Li, C.-B.; Xu, X.-L.; Zhou, G.-H.; Cao, J.-X. iTRAQ-based quantitative proteomic characterizes the salting exudates of Jinhua ham during the salting process. Food Control 2019, 100, 189–197. [Google Scholar] [CrossRef]
- Zhou, C.-Y.; Xia, Q.; He, J.; Sun, Y.-Y.; Dang, Y.-L.; Zhou, G.-H.; Geng, F.; Pan, D.-D.; Cao, J.-X. Insights into ultrasonic treatment on the mechanism of proteolysis and taste improvement of defective dry-cured ham. Food Chem. 2022, 388, 133059. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.-Y.; Wu, J.-Q.; Tang, C.-B.; Li, G.; Dai, C.; Bai, Y.; Li, C.-B.; Xu, X.-L.; Zhou, G.-H.; Cao, J.-X. Comparing the proteomic profile of proteins and the sensory characteristics in Jinhua ham with different processing procedures. Food Control 2019, 106, 106694. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xu, X.; Zhou, G. The roles of the actin-myosin interaction and proteolysis in tenderization during the aging of chicken muscle. Poult. Sci. 2012, 91, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Bhat, Z.F.; Morton, J.D.; Mason, S.L.; Bekhit, A.E.D.A. Role of calpain system in meat tenderness: A review. Food Sci. Hum. Wellness 2018, 7, 196–204. [Google Scholar] [CrossRef]
- Kęska, P.; Stadnik, J. Taste-active peptides and amino acids of pork meat as components of dry-cured meat products: An in-silico study. J. Sens. Stud. 2017, 32, 12301–12311. [Google Scholar] [CrossRef]
- Mora, L.; Sentandreu, M.A.; Koistinen, K.M.; Fraser, P.D.; Toldrá, F.; Bramley, P.M. Naturally generated small peptides derived from myofibrillar proteins in Serrano dry-cured ham. J. Agric. Food Chem. 2009, 57, 3228–3234. [Google Scholar] [CrossRef]
- Lopez-Pedrouso, M.; Lorenzo, J.M.; Perez-Ciria, L.; Ripoll, G.; Latorre, M.A.; Franco, D. A proteomic approach for in-depth characterization and understanding the impact of immunocastration on dry-cured ham of male and female pigs. Food Res. Int. 2022, 154, 111020. [Google Scholar] [CrossRef]
- Juan, E.Y.T.; Li, W.J.; Jhang, J.H.; Chiu, C.H. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 2004, 13, 1402–1406. [Google Scholar]
- Zhang, B.; Mao, J.L.; Yao, H.; Aubourg, S.P. Label-free based proteomics analysis of protein changes in frozen whiteleg shrimp (Litopenaeus vannamei) pre-soaked with sodium trimetaphosphate. Food Res. Int. 2020, 137, 109455. [Google Scholar] [CrossRef]
- Minoru, K.; Susumu, G.; Yoko, S.; Miho, F.; Mao, T. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
- Zhou, C.-Y.; Bai, Y.; Wang, C.; Li, C.-B.; Xu, X.-L.; Pan, D.-D.; Cao, J.-X.; Zhou, G.-H. 1H NMR-based metabolomics and sensory evaluation characterize taste substances of Jinhua ham with traditional and modern processing procedures. Food Control 2021, 126, 107873. [Google Scholar] [CrossRef]
- Arroyo, J.D.; Jourdain, A.A.; Calvo, S.E.; Ballarano, C.A.; Doench, J.G.; Root, D.E.; Mootha, V.K. A Genome-wide CRISPR Death Screen Identifies Genes Essential for Oxidative Phosphorylation. Cell Metab. 2016, 24, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Zequan, X.; Yonggang, S.; Guangjuan, L.; Shijun, X.; Li, Z.; Mingrui, Z.; Yanli, X.; Zirong, W. Proteomics analysis as an approach to understand the formation of pale, soft, and exudative (PSE) pork. Meat Sci. 2021, 177, 108353. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Huang, C.; Blecker, C.; Chen, L.; Xiang, C.; Zheng, X.; Wang, Z.; Zhang, D. Integrating identification and targeted proteomics to discover the potential indicators of postmortem lamb meat quality. Meat Sci. 2023, 199, 109126. [Google Scholar] [CrossRef] [PubMed]
- Poleti, M.D.; Moncau, C.T.; Silva-Vignato, B.; Rosa, A.F.; Lobo, A.R.; Cataldi, T.R.; Negrao, J.A.; Silva, S.L.; Eler, J.P.; de Carvalho Balieiro, J.C. Label-free quantitative proteomic analysis reveals muscle contraction and metabolism proteins linked to ultimate pH in bovine skeletal muscle. Meat Sci. 2018, 145, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Wang, S.-W.; Zhao, B.; Zhang, S.-L.; Zang, M.-W.; Wu, Q.-R.; Li, S.; Qiao, X.-L. Label-free proteomic strategy to identify proteins associated with quality properties in sauced beef processing. Food Biosci. 2021, 42, 101163. [Google Scholar] [CrossRef]
- Benet, I.; Guàrdia, M.D.; Ibañez, C.; Solà, J.; Arnau, J.; Roura, E. Analysis of SPME or SBSE extracted volatile compounds from cooked cured pork ham differing in intramuscular fat profiles. LWT-Food Sci. Technol. 2015, 60, 393–399. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Z.; Ji, L.; Zhang, J.; Zhao, Z.; Zhang, R.; Bai, T.; Hou, B.; Wang, W. Flavor Composition and Microbial Community Structure of Mianning Ham. Front. Microbiol. 2020, 11, 623775. [Google Scholar] [CrossRef]
- Toldrá, F. The role of muscle enzymes in dry-cured meat products with different drying conditions. Trends Food Sci. Technol. 2006, 17, 164–168. [Google Scholar] [CrossRef]
- Scheffler, T.L.; Matarneh, S.K.; England, E.M.; Gerrard, D.E. Mitochondria influence postmortem metabolism and pH in an in vitro model. Meat Sci. 2015, 110, 118–125. [Google Scholar] [CrossRef]
- Ramos, P.M.; Wright, S.A.; Delgado, E.F.; van Santen, E.; Johnson, D.D.; Scheffler, J.M.; Elzo, M.A.; Carr, C.C.; Scheffler, T.L. Resistance to pH decline and slower calpain-1 autolysis are associated with higher energy availability early postmortem in Bos taurus indicus cattle. Meat Sci. 2020, 159, 107925. [Google Scholar] [CrossRef]
- Li, C.-Y.; Lee, J.-S.; Ko, Y.-G.; Kim, J.-I.; Seo, J.-S. Heat Shock Protein 70 Inhibits Apoptosis Downstream of Cytochrome c Release and Upstream of Caspase-3 Activation. J. Biol. Chem. 2000, 275, 25665–25671. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein ID | Gene ID | Protein Name | Intensity (Log10 P) | FC (FH/LH) | p Value | |
|---|---|---|---|---|---|---|
| FH | LH | |||||
| M3VH45 | ANXA6 | Annexin | 5.95 ± 0.12 | 4.75 ± 0.52 | 10.32 | 0.01 |
| F1KQF6 | Glutamyl-tRNA synthetase (Fragment) | 5.48 ± 0.04 | 4.49 ± 0.17 | 9.25 | 0.00 | |
| A0A5G2QQP3 | Histone H2A | 6.51 ± 0.16 | 5.55 ± 0.23 | 8.84 | 0.02 | |
| F1LI18 | Actin-2 (Fragment) | 6.50 ± 0.06 | 5.82 ± 0.14 | 4.61 | 0.00 | |
| L8B0W4 | IGHG | IgG heavy chain | 6.27 ± 0.18 | 5.64 ± 0.17 | 4.27 | 0.05 |
| A0A286XHM4 | TPM1 | Tropomyosin 1 | 6.84 ± 0.10 | 6.23 ± 0.18 | 3.86 | 0.01 |
| Q8MKF3 | TPM2 | Beta-tropomyosin (Fragment) | 7.11 ± 0.07 | 6.48 ± 0.29 | 3.69 | 0.00 |
| A0A5G2QN66 | P4HB | Protein disulfide-isomerase | 4.82 ± 0.14 | 4.19 ± 0.33 | 3.52 | 0.03 |
| A0A4X1SRU3 | PSMC4 | 26S proteasome AAA-ATPase subunit RPT3 | 5.85 ± 0.14 | 5.25 ± 0.31 | 3.49 | 0.03 |
| A0A5G2QYN3 | SYNPO | Synaptopodin | 5.31 ± 0.07 | 4.74 ± 0.24 | 3.37 | 0.00 |
| F1RYZ5 | RPL10A | Ribosomal protein | 5.28 ± 0.05 | 4.74 ± 0.15 | 3.36 | 0.00 |
| A0A286Y2C2 | PPP3R1 | Protein phosphatase 3 regulatory subunit B, alpha | 5.47 ± 0.09 | 5.00 ± 0.11 | 2.92 | 0.02 |
| A0A0H5ANC0 | OGN | Mimecan | 5.70 ± 0.08 | 5.23 ± 0.11 | 2.90 | 0.01 |
| Q2EN79 | UQCR10 | Ubiquinol-cytochrome c reductase complex | 6.03 ± 0.18 | 5.58 ± 0.21 | 2.79 | 0.05 |
| A0A5G2QN78 | GRB2 | Growth factor receptor bound protein 2 | 5.39 ± 0.11 | 4.94 ± 0.17 | 2.73 | 0.02 |
| F1SRY6 | PLCD4 | Phosphoinositide phospholipase C | 5.88 ± 0.14 | 5.41 ± 0.28 | 2.70 | 0.03 |
| F1SKJ0 | ARIH2 | RBR-type E3 ubiquitin transferase | 5.68 ± 0.03 | 5.20 ± 0.26 | 2.66 | 0.04 |
| F1SUE4 | ASPN | Asporin | 6.12 ± 0.12 | 5.66 ± 0.23 | 2.63 | 0.04 |
| Q4PS85 | MYOZ1 | Myozenin-1 | 7.66 ± 0.04 | 7.25 ± 0.07 | 2.58 | 0.00 |
| A0A480XDR9 | Biglycan (Fragment) | 6.58 ± 0.11 | 6.15 ± 0.22 | 2.56 | 0.02 | |
| A0A4X1TG89 | ALDH6A1 | Aldedh domain-containing protein | 5.36 ± 0.04 | 4.91 ± 0.26 | 2.51 | 0.02 |
| A0A4X1T3Z4 | DNASE1L1 | Deoxyribonuclease | 5.62 ± 0.07 | 5.22 ± 0.12 | 2.42 | 0.02 |
| A0A480WUT2 | Endophilin-B1 isoform 1 | 5.01 ± 0.07 | 4.60 ± 0.21 | 2.37 | 0.01 | |
| F1S227 | PTRH2 | Aminoacyl-tRNA hydrolase | 5.70 ± 0.11 | 5.32 ± 0.16 | 2.36 | 0.02 |
| A0A480K0C0 | Collagen alpha-3(VI) chain isoform X4 | 5.62 ± 0.01 | 3.48 ± 3.02 | 2.32 | 0.02 | |
| A0A2I6SB80 | Thy-1 cell surface antigen (Fragment) | 5.53 ± 0.12 | 5.19 ± 0.17 | 2.13 | 0.04 | |
| A0A4X1U9Z4 | NPM1 | Nucleoplasmin domain-containing protein | 5.09 ± 0.03 | 4.78 ± 0.09 | 2.02 | 0.00 |
| A0A287AY74 | FBP2 | Fructose-bisphosphatase | 7.53 ± 0.02 | 7.22 ± 0.09 | 2.02 | 0.00 |
| Q7YS28 | Pyruvate carboxylase | 5.56 ± 0.10 | 5.24 ± 0.18 | 2.01 | 0.05 | |
| A0A2K6CXN1 | NEB | SH3 domain-containing protein | 5.85 ± 0.10 | 5.54 ± 0.14 | 1.96 | 0.05 |
| A0A5G2Q828 | PNPO | Pyridoxal 5-phosphate synthase | 5.79 ± 0.02 | 5.48 ± 0.16 | 1.96 | 0.01 |
| A0A286ZKJ2 | PFKM | ATP-dependent 6-phosphofructokinase | 7.88 ± 0.07 | 7.60 ± 0.01 | 1.92 | 0.01 |
| I3LVF5 | PA2G4 | Peptidase_M24 domain-containing protein | 5.32 ± 0.07 | 5.02 ± 0.19 | 1.91 | 0.03 |
| F1SQ01 | PRDX4 | Peroxiredoxin-4 | 6.16 ± 0.05 | 5.85 ± 0.20 | 1.91 | 0.02 |
| A0A0B8RVD8 | VIM | Vimentin | 6.85 ± 0.03 | 6.55 ± 0.17 | 1.90 | 0.01 |
| K7GM40 | APOA1 | Apolipoprotein A-I | 5.72 ± 0.07 | 5.41 ± 0.20 | 1.89 | 0.05 |
| A0A480WWH8 | Basement membrane-specific heparan sulfate proteoglycan core protein isoform X4 | 6.50 ± 0.02 | 6.22 ± 0.02 | 1.88 | 0.00 | |
| F1S073 | ANXA2 | Annexin | 7.08 ± 0.02 | 6.80 ± 0.11 | 1.87 | 0.01 |
| A0A480JV40 | Lamin isoform A | 6.82 ± 0.05 | 6.53 ± 0.15 | 1.86 | 0.02 | |
| A0A4X1UHN0 | IF rod domain-containing protein | 6.75 ± 0.12 | 6.50 ± 0.06 | 1.82 | 0.05 | |
| A0A4X1UEP5 | HSPA5 | 78 kDa glucose-regulated protein | 6.41 ± 0.09 | 6.16 ± 0.07 | 1.79 | 0.03 |
| A0A481CHK3 | Alpha-actinin-4 isoform 1 | 6.82 ± 0.03 | 6.53 ± 0.15 | 1.79 | 0.03 | |
| A0A2K6D3R9 | DES | IF rod domain-containing protein | 7.91 ± 0.10 | 7.67 ± 0.10 | 1.77 | 0.04 |
| A0A5G2QUU1 | FGG | Fibrinogen gamma chain | 5.96 ± 0.09 | 5.72 ± 0.06 | 1.73 | 0.02 |
| F1SH96 | ITIH1 | Inter-alpha-trypsin inhibitor heavy chain H1 | 5.62 ± 0.01 | 5.39 ± 0.06 | 1.71 | 0.00 |
| Q6J267 | Galectin (Fragment) | 7.42 ± 0.03 | 7.20 ± 0.05 | 1.66 | 0.00 | |
| A0A480Z467 | Neuroblast differentiation-associated protein AHNAK isoform X1 | 6.03 ± 0.08 | 5.81 ± 0.08 | 1.66 | 0.03 | |
| Q6QAQ0 | 40S ribosomal protein S20 (Fragment) | 5.31 ± 0.05 | 5.08 ± 0.11 | 1.65 | 0.02 | |
| A0A4X1SJ46 | ANXA11 | Annexin | 6.20 ± 0.09 | 6.00 ± 0.05 | 1.60 | 0.04 |
| A0A480S836 | Glucose-6-phosphate isomerase | 7.86 ± 0.03 | 7.65 ± 0.12 | 1.59 | 0.02 | |
| A0A287A0V9 | PHKB | Phosphorylase b kinase regulatory subunit | 6.13 ± 0.03 | 5.94 ± 0.04 | 1.54 | 0.00 |
| A1XQT6 | MYL1 | MLC1f | 8.11 ± 0.06 | 7.93 ± 0.06 | 1.52 | 0.03 |
| F1RX45 | DIP2C | Disco interacting protein 2 homolog C | 4.53 ± 0.06 | 4.71 ± 0.04 | 0.66 | 0.02 |
| A0A480ZTV3 | Endonuclease domain-containing 1 protein | 5.27 ± 0.11 | 5.47 ± 0.02 | 0.65 | 0.02 | |
| B1Q039 | CRYAB | Alpha(B)-crystallin | 7.42 ± 0.06 | 7.61 ± 0.07 | 0.65 | 0.03 |
| A0A4X1VII3 | ATP6V1B2 | ATP-synt_ab domain-containing protein | 3.06 ± 0.02 | 4.78 ± 0.02 | 0.65 | 0.01 |
| A0A480SHW2 | Myosin-7 | 8.57 ± 0.06 | 8.76 ± 0.01 | 0.65 | 0.00 | |
| F1RKW8 | PSMD11 | Proteasome 26S subunit, non-ATPase 11 | 5.77 ± 0.07 | 5.96 ± 0.05 | 0.64 | 0.01 |
| F1SKM0 | UQCRC1 | Ubiquinol-cytochrome c reductase core protein 1 | 6.76 ± 0.05 | 6.97 ± 0.06 | 0.62 | 0.01 |
| A0A2K6CCF8 | UCHL3 | Ubiquitin carboxyl-terminal hydrolase | 5.91 ± 0.06 | 6.13 ± 0.09 | 0.60 | 0.04 |
| F1SA50 | HNRNPM | Heterogeneous nuclear ribonucleoprotein M | 4.73 ± 0.21 | 5.02 ± 0.01 | 0.55 | 0.05 |
| A0A5S6HSJ6 | ATP2A2 | Calcium-transporting ATPase | 7.06 ± 0.03 | 7.33 ± 0.08 | 0.53 | 0.01 |
| B9ZUV0 | COII | Cytochrome c oxidase subunit 2 | 6.60 ± 0.15 | 6.90 ± 0.06 | 0.51 | 0.02 |
| A0A4X1SL20 | MYH2 | Myosin-2 | 7.92 ± 0.05 | 8.22 ± 0.01 | 0.51 | 0.00 |
| F1RSZ3 | ACSL1 | Acyl-CoA synthetase long chain family member 1 | 6.32 ± 0.25 | 6.65 ± 0.05 | 0.51 | 0.04 |
| A0A5G2QFC2 | MDH1 | Malate dehydrogenase | 6.35 ± 0.12 | 6.66 ± 0.07 | 0.50 | 0.01 |
| A0A287AP18 | NRAP | Nebulin related anchoring protein | 5.62 ± 0.15 | 5.94 ± 0.01 | 0.50 | 0.01 |
| F1RHN3 | COMT | Catechol O-methyltransferase | 5.30 ± 0.19 | 5.64 ± 0.07 | 0.48 | 0.05 |
| A0A1B2TT55 | GOT1 | Aspartate aminotransferase | 7.07 ± 0.10 | 7.42 ± 0.04 | 0.46 | 0.00 |
| B0F9U1 | Alpha 1 S haptoglobin (Fragment) | 5.04 ± 0.15 | 5.41 ± 0.12 | 0.44 | 0.04 | |
| A0A4X1V532 | NDUFB7 | Complex I-B18 | 4.39 ± 0.09 | 4.76 ± 0.07 | 0.44 | 0.05 |
| A0A4X1SLC1 | Protein-serine/threonine phosphatase | 4.27 ± 0.12 | 4.65 ± 0.02 | 0.42 | 0.01 | |
| A0A4X1SM67 | MAP2K6 | Protein kinase domain-containing protein | 5.64 ± 0.13 | 6.06 ± 0.10 | 0.39 | 0.02 |
| A0A0B8RZ32 | STIP1 | Stress-induced phosphoprotein 1 | 5.41 ± 0.11 | 5.84 ± 0.08 | 0.37 | 0.01 |
| I3LV35 | CPT1B | Carnitine O-palmitoyltransferase | 5.31 ± 0.14 | 5.74 ± 0.14 | 0.37 | 0.04 |
| A0A480JX12 | Integrin beta | 5.38 ± 0.56 | 6.00 ± 0.06 | 0.36 | 0.03 | |
| Q8SPJ9 | COX7A1 | Cytochrome c oxidase subunit 7A1, mitochondrial | 6.09 ± 0.08 | 6.53 ± 0.13 | 0.36 | 0.01 |
| P10775 | RNH1 | Ribonuclease inhibitor | 5.67 ± 0.10 | 6.12 ± 0.10 | 0.35 | 0.01 |
| F1RG17 | NME3 | Nucleoside diphosphate kinase | 4.88 ± 0.70 | 5.61 ± 0.07 | 0.32 | 0.02 |
| A0A2K6CDX4 | PGAM2 | Phosphoglycerate mutase | 5.62 ± 0.39 | 6.25 ± 0.05 | 0.28 | 0.02 |
| A0A2K6ED17 | CAMK2D | Calcium/calmodulin-dependent protein kinase | 5.08 ± 0.66 | 5.97 ± 0.07 | 0.28 | 0.04 |
| A0A4X1SNG6 | APOBEC2 | CMP/dCMP-type deaminase domain-containing protein | 5.96 ± 0.29 | 6.63 ± 0.09 | 0.25 | 0.01 |
| A0A5G2R4Z6 | LMCD1 | LIM and cysteine-rich domains protein 1 | 5.80 ± 0.17 | 6.45 ± 0.11 | 0.23 | 0.01 |
| A0A480HZP7 | Protein flightless-1 homolog isoform 1 | 4.26 ± 0.11 | 4.93 ± 0.15 | 0.21 | 0.02 | |
| A0A286XNT8 | TPR_REGION domain-containing protein | 4.40 ± 0.52 | 5.24 ± 0.08 | 0.20 | 0.01 | |
| F1SHD6 | EEF1B2 | Elongation factor 1-beta | 4.80 ± 0.89 | 5.89 ± 0.11 | 0.18 | 0.01 |
| A0A480TZB5 | Heat shock 70 kDa protein 4 | 5.02 ± 0.35 | 5.91 ± 0.12 | 0.16 | 0.01 | |
| A0A480MYX8 | Laminin subunit alpha-5 isoform X1 | 4.41 ± 0.25 | 5.27 ± 0.23 | 0.14 | 0.04 | |
| A0A5G2QN18 | SUGT1 | SGT1 homolog, MIS12 kinetochore complex assembly cochaperone | 4.69 ± 0.84 | 5.89 ± 0.15 | 0.14 | 0.02 |
| A0A481D2X2 | Protein-serine/threonine kinase | 3.87 ± 0.62 | 5.17 ± 0.19 | 0.07 | 0.01 | |
| Q6V9B4 | APM1 | Adiponectin (Fragment) | 3.74 ± 0.26 | 5.59 ± 0.03 | 0.02 | 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Q.; Xie, R.; Wu, X.; Zhao, K.; Li, H.; Tang, H.; Du, H.; Peng, X.; Chen, L.; Zhang, J. Insights into the Protein Differentiation Mechanism between Jinhua Fatty Ham and Lean Ham through Label-Free Proteomics. Foods 2023, 12, 4348. https://doi.org/10.3390/foods12234348
Huang Q, Xie R, Wu X, Zhao K, Li H, Tang H, Du H, Peng X, Chen L, Zhang J. Insights into the Protein Differentiation Mechanism between Jinhua Fatty Ham and Lean Ham through Label-Free Proteomics. Foods. 2023; 12(23):4348. https://doi.org/10.3390/foods12234348
Chicago/Turabian StyleHuang, Qicheng, Ruoyu Xie, Xiaoli Wu, Ke Zhao, Huanhuan Li, Honggang Tang, Hongying Du, Xinyan Peng, Lihong Chen, and Jin Zhang. 2023. "Insights into the Protein Differentiation Mechanism between Jinhua Fatty Ham and Lean Ham through Label-Free Proteomics" Foods 12, no. 23: 4348. https://doi.org/10.3390/foods12234348
APA StyleHuang, Q., Xie, R., Wu, X., Zhao, K., Li, H., Tang, H., Du, H., Peng, X., Chen, L., & Zhang, J. (2023). Insights into the Protein Differentiation Mechanism between Jinhua Fatty Ham and Lean Ham through Label-Free Proteomics. Foods, 12(23), 4348. https://doi.org/10.3390/foods12234348