
Selective Elucidation of Living Microbial Communities in Fermented Grains of Chinese Baijiu: Development of a Technique Integrating Propidium Monoazide Probe Pretreatment and Amplicon Sequencing

 , and
, and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Suspension Preparation of Fermented Grain Samples
2.2. Preparation of PMA Working Solution
2.3. Preparation of Positive Control Group
2.4. Establishment of PMA Pre-Treatment Method for Fermented Grains
2.4.1. Selection of Fermented Grain Samples for Optimization of the PMA Pre-Treatment Protocol
2.4.2. Optimization of PMA Concentration
2.4.3. Optimization of Dark Incubation Time
2.4.4. Optimization of Exposure Time
2.5. Bacterial Strains, Plasmids and Cultivation
2.6. Amplicon Sequencing
2.7. qPCR and PCR Reaction System and Procedure
2.8. Statistical Analyses
3. Results
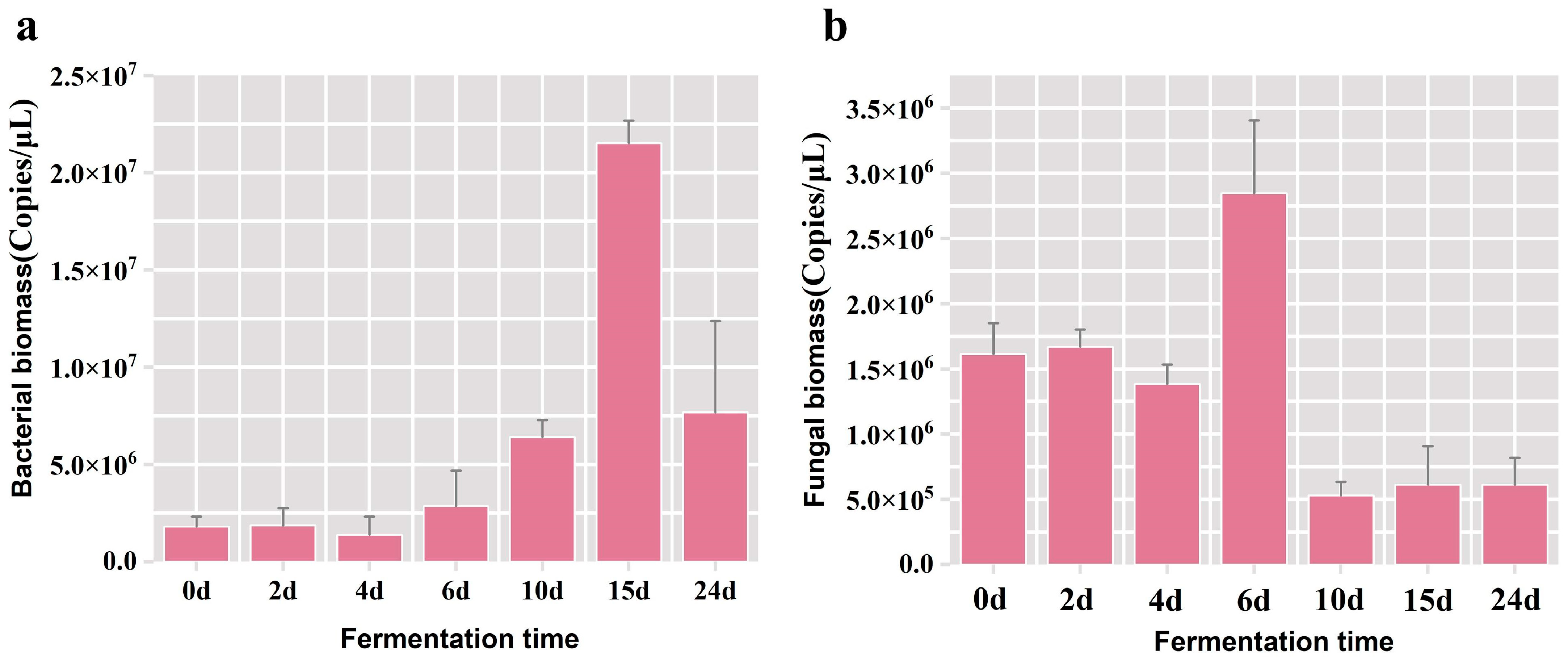
3.1. Selection of Fermented Grains for Optimization
3.2. Optimization of PMA Pre-Treatment Conditions for Fermented Grains
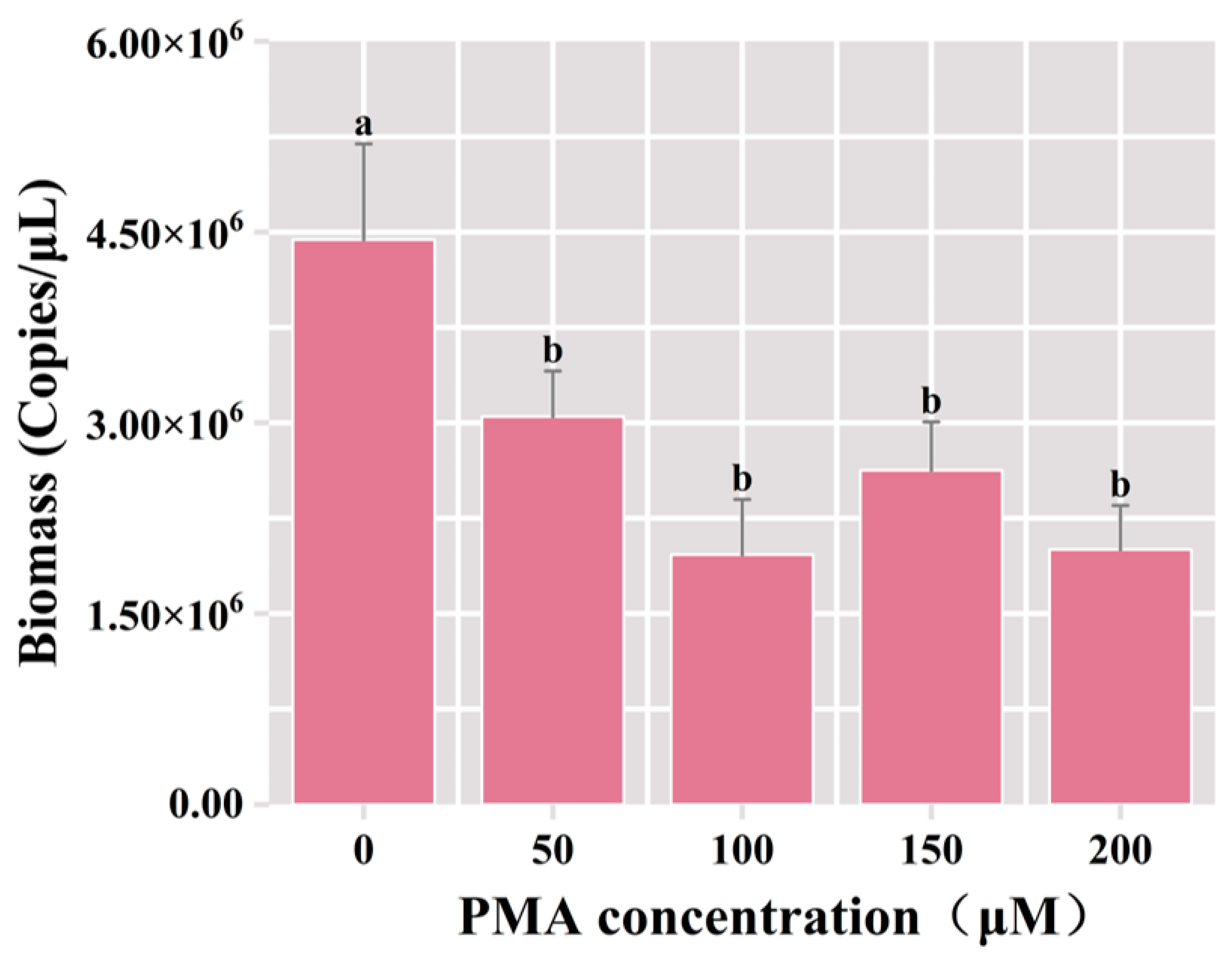
3.2.1. Optimization of PMA Concentration
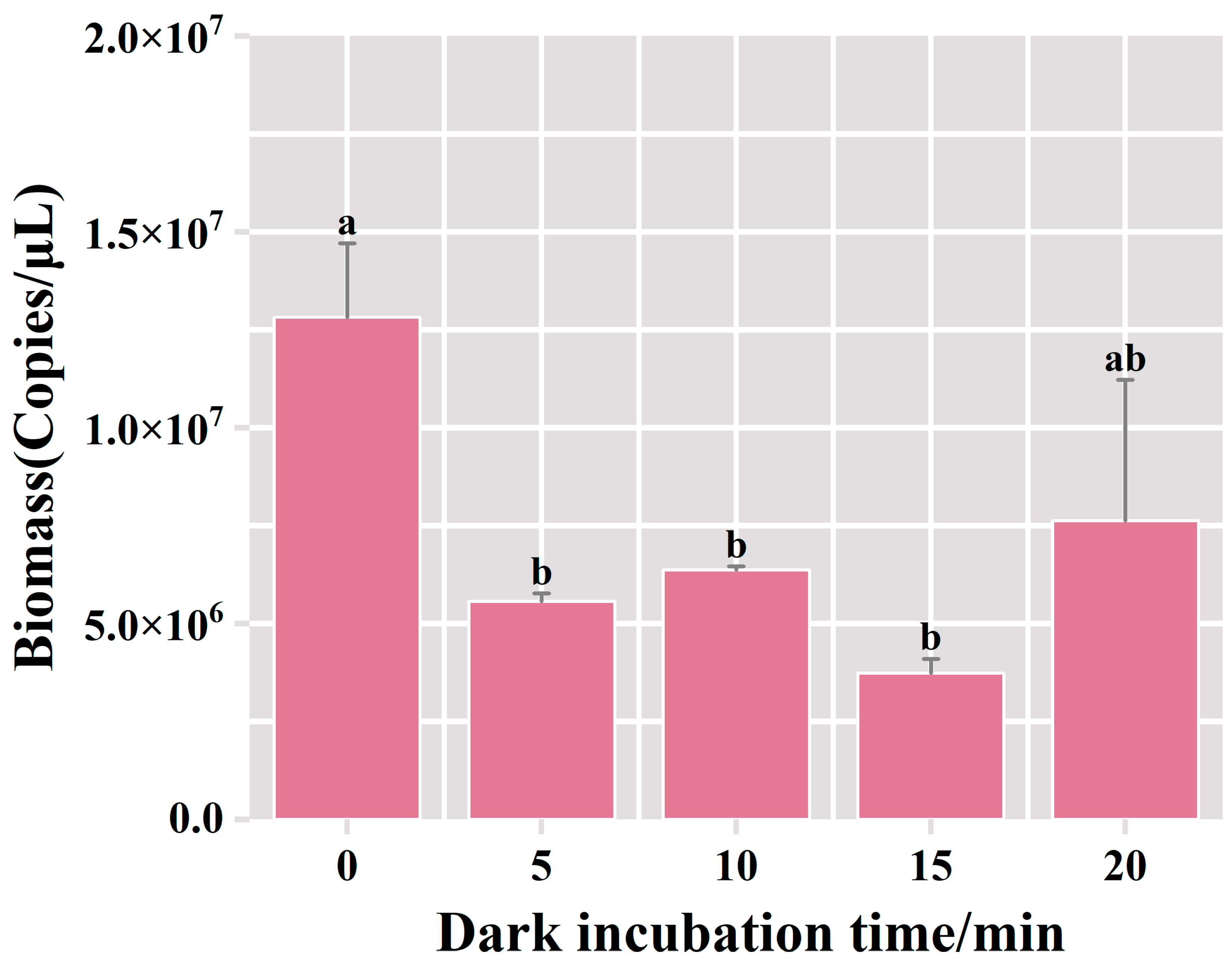
3.2.2. Optimization of Duration for Incubation in Darkness
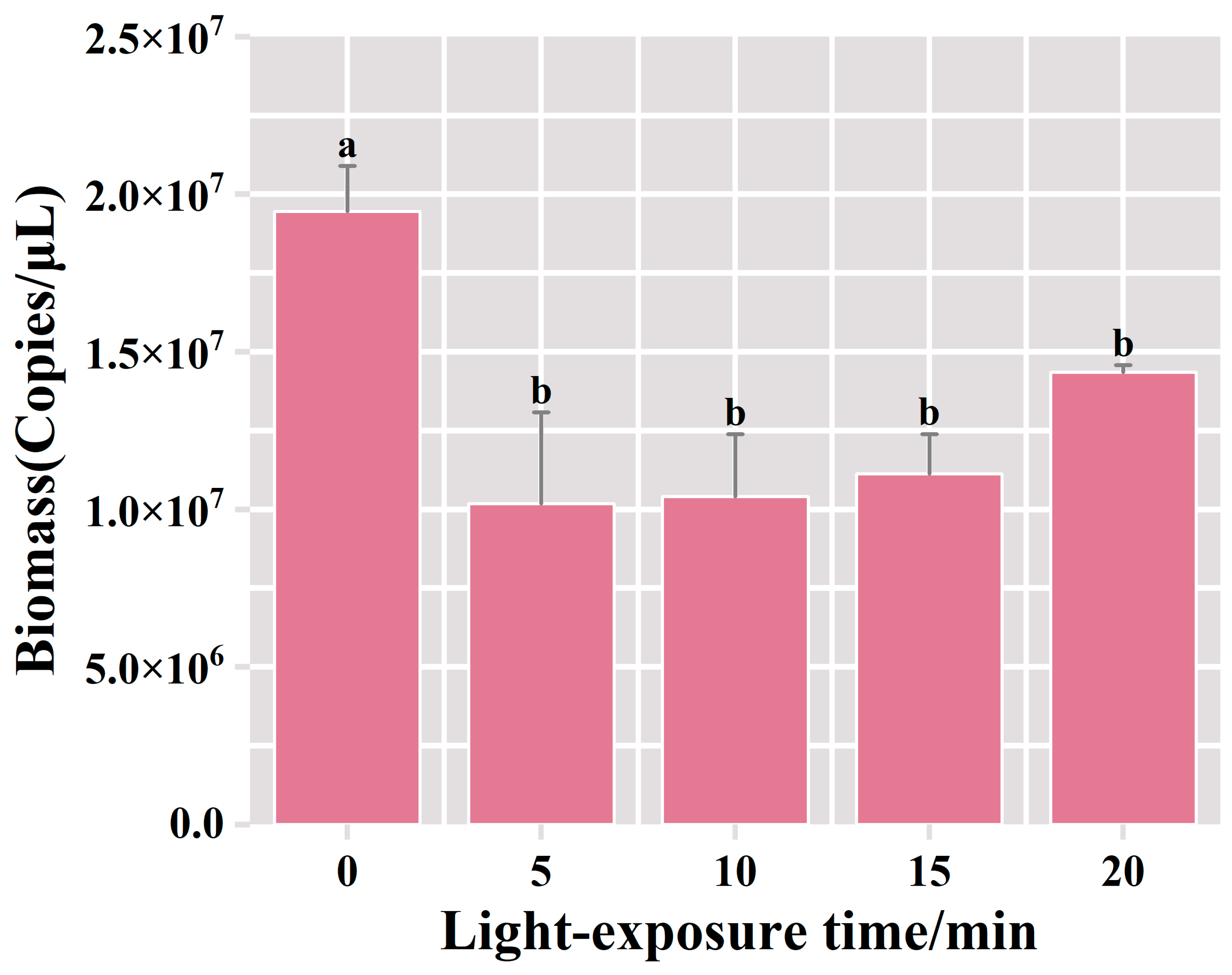
3.2.3. Optimization of Exposure Time
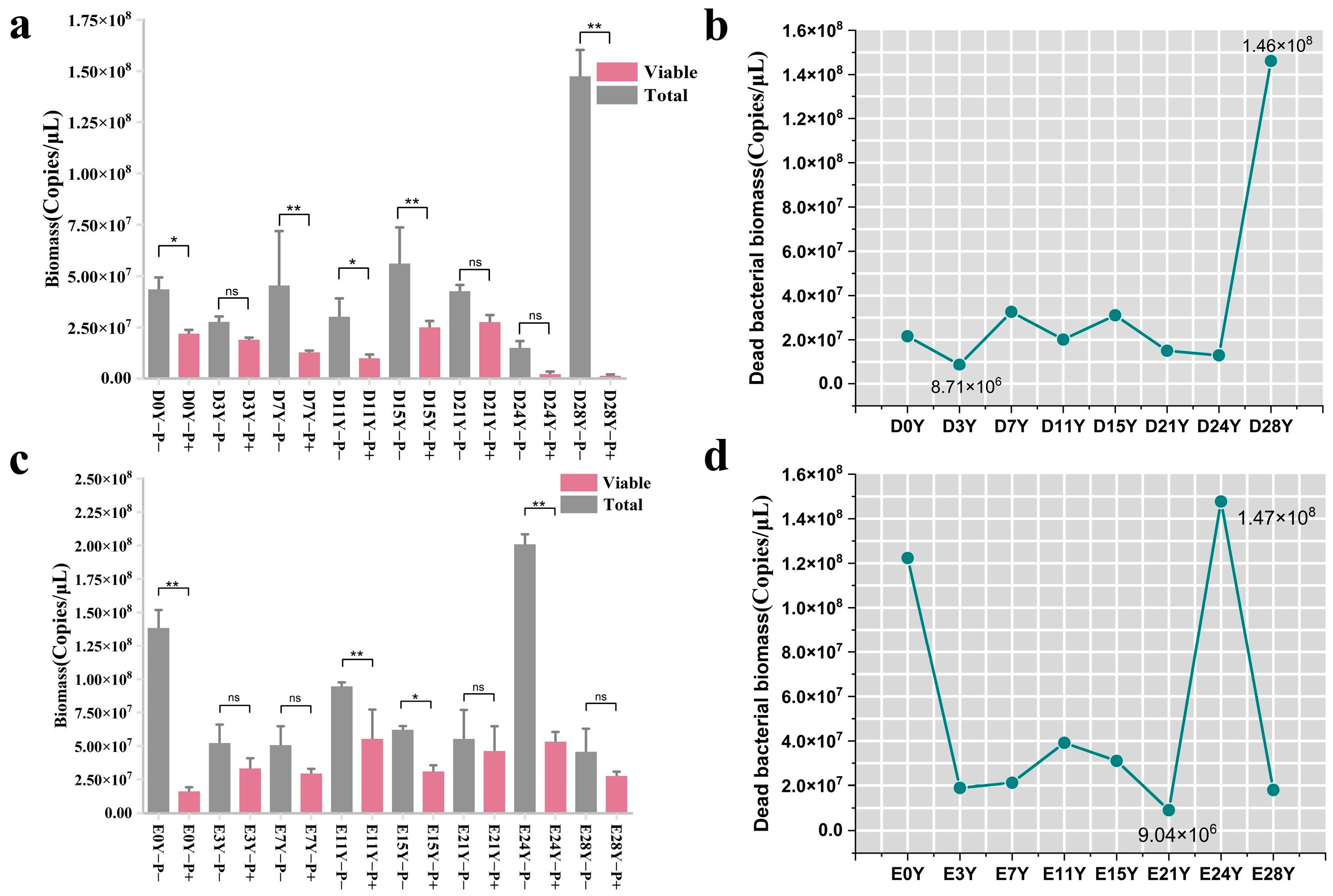
3.3. Assessment of the Binding Efficiency of PMA with the DNA of Deceased Bacteria in Fermented Grains under Optimal Treatment Conditions
3.4. Evaluation of the Efficacy in Deciphering the Living Microbial Community Profile in Fermented Grains through the Coupling of PMA Pretreatment Technology with Amplicon Sequencing
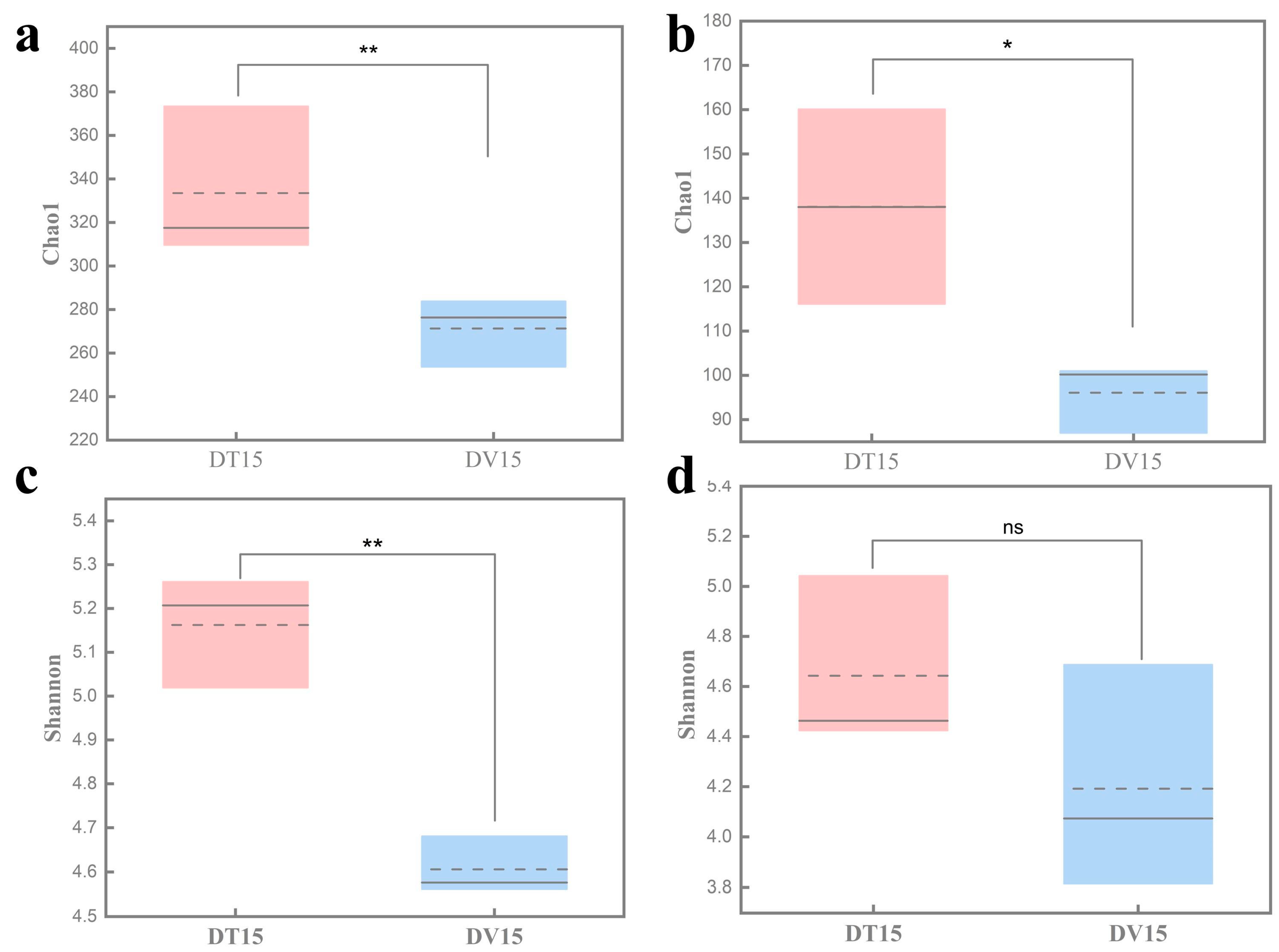
3.4.1. The α-Diversity of the Active Bacterial Community in Fermented Grains Exhibited a Markedly Reduced Level Compared to the Total Bacterial Community
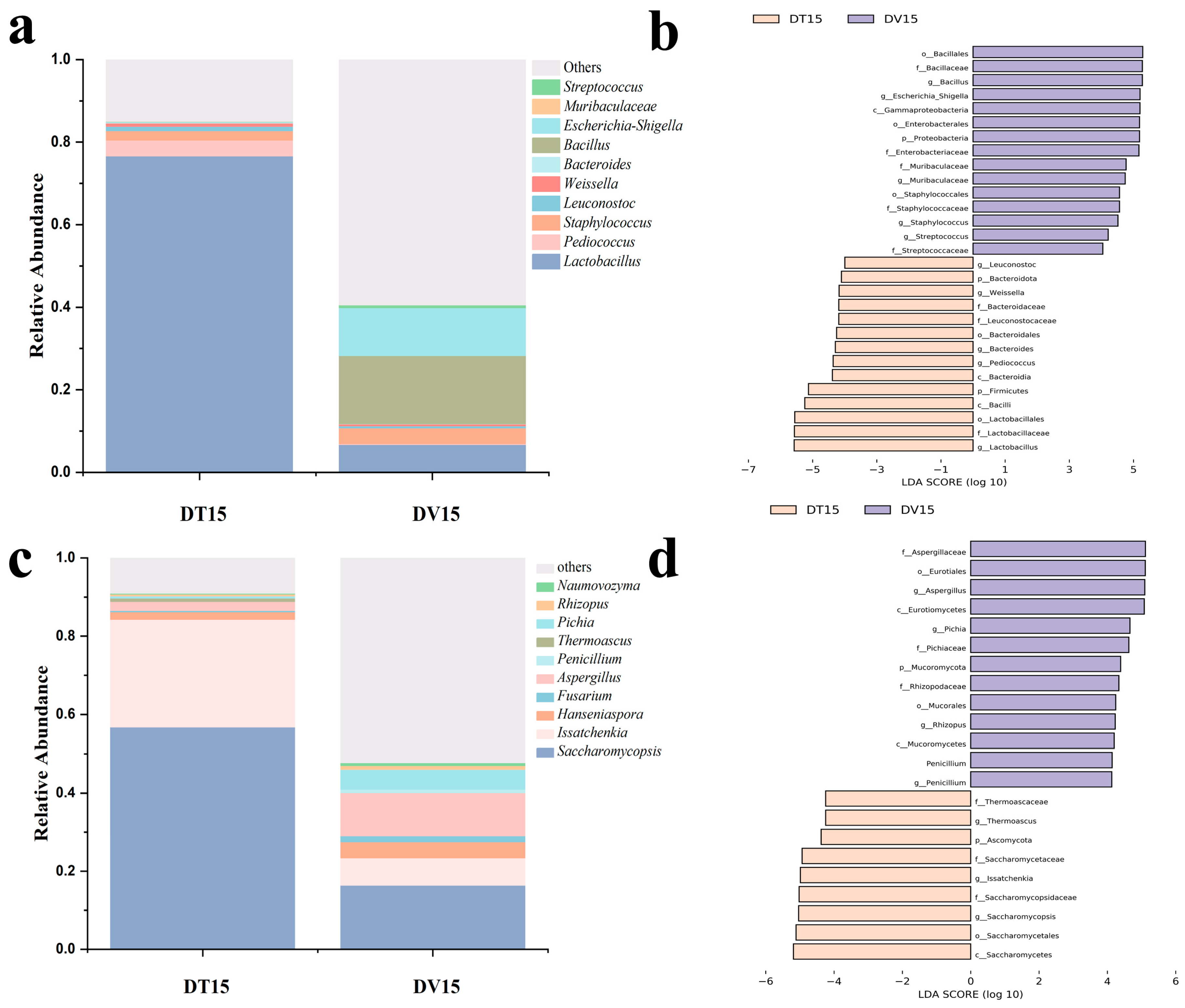
3.4.2. Differences in Bacterial and Fungal Microbial Structure Composition of the DT and DV Groups
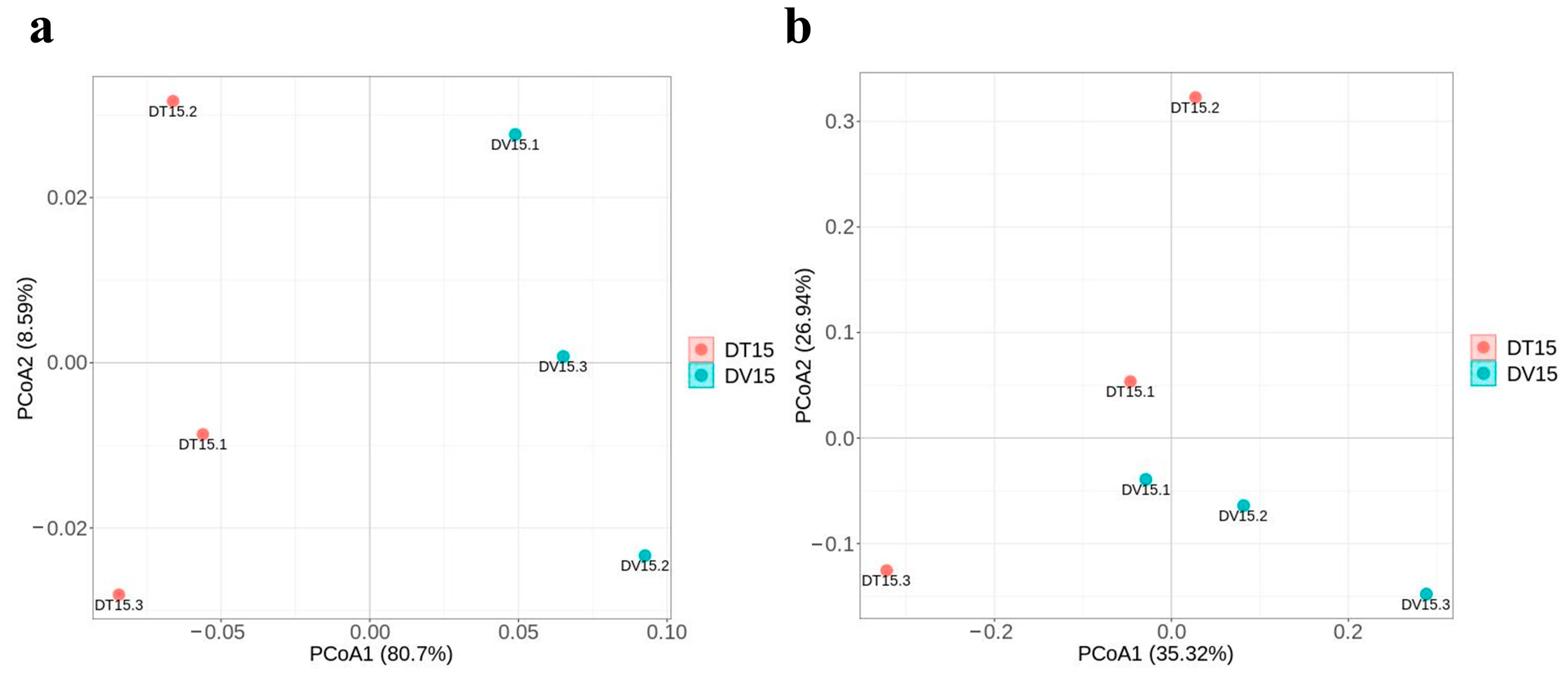
3.4.3. β-Diversity of the Bacterial and Fungal Community Revealed a Clear Separation between the DT and DV Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dimidi, E.; Cox, S.R.; Rossi, M.; Whelan, K. Fermented Foods: Definitions and Characteristics, Impact on the Gut Microbiota and Effects on Gastrointestinal Health and Disease. Nutrients 2019, 11, 1806. [Google Scholar] [CrossRef] [PubMed]
- Du, R.B.; Ren, C.; Wu, Q.; Xu, Y. The ecological fermentation technology: Principle and its applications. Food Ferment. Ind. 2021, 47, 266–275. [Google Scholar] [CrossRef]
- Chen, J.; Wang, C.; Zhu, Q.; Zhang, J. Research Status and Application Prospect of Frontier Technology of Traditional Fermented Food in China. J. Food Sci. Technol. 2021, 39, 1–7. [Google Scholar]
- Liu, A.; Ou, Y.; Shu, H.; Mou, T.; Li, Q.; Li, J.; Hu, K.; Chen, S.; He, L.; Zhou, J.; et al. Exploring the role of Sichuan Baoning vinegar microbiota and the association with volatile flavor compounds at different fermentation depths. Front. Microbiol. 2023, 14, 1135912. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Kim, S.H.; Jeong, W.S.; Kim, S.Y.; Yeo, S.H. Microbiome Analysis of Traditional Grain Vinegar Produced under Different Fermentation Conditions in Various Regions in Korea. Foods 2022, 11, 3573. [Google Scholar] [CrossRef]
- Jiang, L.; Xian, S.; Liu, X.; Shen, G.; Zhang, Z.; Hou, X.; Chen, A. Metagenomic Study on Chinese Homemade Paocai: The Effects of Raw Materials and Fermentation Periods on the Microbial Ecology and Volatile Components. Foods 2021, 11, 62. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Du, H.; Zhang, H.; Fang, C.; Jin, G.; Chen, S.; Xu, Y. Geographically Associated Fungus-Bacterium Interactions Contribute to the Formation of Geography-Dependent Flavor during High-Complexity Spontaneous Fermentation. Microbiol. Spectr. 2022, 10, e0184422. [Google Scholar] [CrossRef] [PubMed]
- Luo, A.; Yang, N.; Yang, J.; Hao, J.; Zhao, J.; Shi, S.; Hu, B. Effects of microbial interspecies relationships and physicochemical parameters on volatile flavors in sorghum-based fermented grains during the fermentation of Shanxi light-flavored liquor. Food Sci. Nutr. 2022, 11, 1452–1462. [Google Scholar] [CrossRef] [PubMed]
- Blagodatskaya, E.; Kuzyakov, Y. Active microorganisms in soil: Critical review of estimation criteria and approaches. Soil Biol. Biochem. 2013, 67, 192–211. [Google Scholar] [CrossRef]
- Kong, L.H. Analysis on Active Microorganisms in Air-Dried Agricultural Soils Based on Propidium Monoazid Incorporating High-Throughput sequencing. Master’s Thesis, Northeast Agricultural University, Harbin, China, 2023. [Google Scholar]
- Roth, P.; Hill-Spanik, K.M.; McCurry, C.; Plante, C. Propidium monoazide-denaturing gradient gel electrophoresis (PMA-DGGE) assay for the characterization of viable diatoms in marine sediments. Diatom Res. 2017, 32, 341–350. [Google Scholar] [CrossRef]
- Hu, X.; Wang, K.; Chen, M.; Fan, J.; Han, S.; Hou, J.; Chi, L.; Liu, Y.; Wei, T. Profiling the composition and metabolic activities of microbial community in fermented grain for the Chinese strong-flavor Baijiu production by using the metatranscriptome, high-throughput 16S rRNA and ITS gene sequencings. Food Res. Int. 2020, 138, 109765. [Google Scholar] [CrossRef] [PubMed]
- Solden, L.; Lloyd, K.; Wrighton, K. The bright side of microbial dark matter: Lessons learned from the uncultivated majority. Curr. Opin. Microbiol. 2016, 31, 217–226. [Google Scholar] [CrossRef] [PubMed]
- An, J.X.; Wu, S.H.; Zeng, L.J.; Hunag, J.X.; Feng, P.X.; Liang, T.L.; Yi, Y. Comparison of different methods to extract total RNA from Saccharomycescerevisiae. China Brew. 2021, 40, 82–86. [Google Scholar]
- Ojala, T.; Häkkinen, A.E.; Kankuri, E.; Kankainen, M. Current concepts, advances, and challenges in deciphering the human microbiota with metatranscriptomics. Trends Genet. 2023, 39, 686–702. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, W.; Ma, Y. Real-time and visual detection of viable Salmonella in milk by a competitive annealing mediated isothermal amplification (CAMP) combined with propidium monoazide (PMA). Anal. Methods 2022, 14, 3773–3779. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Shu, Y.; Xia, W.; Xu, R.; Chen, Y. Modified PMA-qPCR Method for Rapid Quantification of Viable Lactobacillus spp. in Fermented Dairy Products. Food Anal. Methods 2021, 14, 1908–1918. [Google Scholar] [CrossRef]
- Ayaka, O.; Mizuki, T.; Kazuha, A.; Ayaka, Y.; Matiur, R.M.; Yasuo, I. Research Note: Detection of Campylobacter spp. in chicken meat using culture methods and quantitative PCR with propidium monoazide. Poult. Sci. 2023, 102, 102883. [Google Scholar]
- Shirasaki, N.; Matsushita, T.; Matsui, Y.; Koriki, S. Suitability of pepper mild mottle virus as a human enteric virus surrogate for assessing the efficacy of thermal or free-chlorine disinfection processes by using infectivity assays and enhanced viability PCR. Water Res. 2020, 186, 116409. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Ji, J.; Li, Y.Y.; Kubota, K. Propidium monoazide-polymerase chain reaction reveals viable microbial community shifts in anaerobic membrane bioreactors treating domestic sewage at low temperature. Bioresour. Technol. 2023, 387, 129564. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Li, X.; Fan, X.; Xu, J.; Liu, Q.; Wu, Z.; Pan, D. PMA-qPCR method for the selective quantitation of viable lactic acid bacteria in fermented milk. Front. Microbiol. 2022, 13, 984506. [Google Scholar] [CrossRef]
- Guo, J.; Wang, W.; Zhao, H.; Luo, Y.; Wan, M.; Li, Y. A new PMA-qPCR method for rapid and accurate detection of viable bacteria and spores of marine-derived Bacillus velezensis B-9987. J. Microbiol. Methods 2022, 199, 106537. [Google Scholar] [CrossRef] [PubMed]
- Ángeles, R.M.d.L.; Anabel, R.R.; Luma, R.R.; Fabiano, D.S.C.; Mariana, C.; Valeria, M.M.; Marcelo, C.; Ramón, V.S. High-pressure processing treatment of beef burgers: Effect on Escherichia coli O157 inactivation evaluated by plate count and PMA-qPCR. J. Food Sci. 2022, 87, 2324–2336. [Google Scholar]
- Liu, H.; Tan, G.; Chen, Q.; Dong, W.; Chen, P.; Cai, K.; Hu, Y.; Zhang, W.; Peng, N.; Liang, Y.; et al. Detection of viable and total fungal community in zaopei of Chinese strong-flavor baijiu using PMA combined with qPCR and HTS based on ITS2 region. BMC Microbiol. 2021, 21, 274. [Google Scholar] [CrossRef] [PubMed]
- Miotto, M.; Barretta, C.; Ossai, S.O.; Silva, H.S.d.; Kist, A.; Vieira, C.R.W.; Parveen, S. Optimization of a propidium monoazide-qPCR method for Escherichia coli quantification in raw seafood. Int. J. Food Microbiol. 2020, 318, 108467. [Google Scholar] [CrossRef] [PubMed]
- Kibbee, R.J.; Örmeci, B. Development of a sensitive and false-positive free PMA-qPCR viability assay to quantify VBNC Escherichia coli and evaluate disinfection performance in wastewater effluent. J. Microbiol. Methods 2017, 132, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Yan, X.; Yang, S.; Chen, F. Screening of Bacillus strains from Luzhou-flavor liquor making for high-yield ethyl hexanoate and low-yield propanol. LWT 2017, 77, 60–66. [Google Scholar] [CrossRef]
- Huang, T. Functional Analysis of Microbial Community of Zhenjiang Aromatic Vinegar and Construction of a Defined Starter for Acitic Acid Production. Master’s Thesis, Jiangnan University, Wuxi, China, 2022. [Google Scholar]
- Xing, Y.; Chen, Y.; Feng, C.; Bao, J.; Li, X.; Jiang, H. Establishment and Application of Real-Time Fluorescence Quantitative PCR Detection Technology for Metschnikowia bicuspidata Disease in Eriocheir sinensis. J. Fungi 2023, 9, 791. [Google Scholar] [CrossRef]
- Hong, B.M. Investigation of Suppressing Anopheles Stephensi Reproduction with RNAi. In Proceedings of the 2nd International Conference on Biological Engineering and Medical Science (ICBioMed 2022), Oxford, UK, 7 November 2022. [Google Scholar]
- Villasante, A.; Ramírez, C.; Rodríguez, H.; Dantagnan, P.; Hernández, A.; Figueroa, E.; Romero, J. Dietary carbohydrate-to-protein ratio influences growth performance, hepatic health and dynamic of gut microbiota in atlantic salmon (Salmo salar). Anim. Nutr. 2022, 10, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Zong, E.; Bo, T.; Dang, L.; Zhang, J.; Li, H.; Lv, N.; He, Y.; Bai, B.; Zhang, J.; Fan, S. Different functions can be provided by low temperature Daqu with different appearance features due to variations in the microbial community structure during fermentation. LWT 2024, 193, 115763. [Google Scholar] [CrossRef]
- Huang, Y.; Li, D.; Mu, Y.; Zhu, Z.; Wu, Y.; Qi, Q.; Mu, Y.; Su, W. Exploring the heterogeneity of community and function and correspondence of “species-enzymes” among three types of Daqu with different fermentation peak-temperature via high-throughput sequencing and metagenomics. Food Res. Int. 2024, 176, 113805. [Google Scholar] [CrossRef]
- Tao, J.L.; LU ZM, W.Z. Detection of the variation of microorganisms in acetic acid fermentation of Zhenjiang aromatic vinegar through real-time quantitative PCR. Food Ferment. Ind. 2013, 39, 156–160. [Google Scholar] [CrossRef]
- Wang, J.; Hao, S.; Ren, Q. Analysis of Bacterial Diversity in Fermented Grains of Baijiu Based on Culturomics and Amplicon Sequencing. Fermentation 2023, 9, 260. [Google Scholar] [CrossRef]
- Chen, X.; Wu, Y.; Zhu, H.; Wang, H.; Lu, H.; Zhang, C.; Li, X.; Xu, Y.; Li, W.; Wang, Y. Turning over fermented grains elevating heap temperature and driving microbial community succession during the heap fermentation of sauce-flavor baijiu. LWT 2022, 172, 114173. [Google Scholar] [CrossRef]
- Zeng, Y.; Wang, Y.; Chen, Q.; Xia, X.; Liu, Q.; Chen, X.; Zhu, B. Dynamics of microbial community structure and enzyme activities during the solid-state fermentation of Forgood Daqu: A starter of Chinese strong flavour Baijiu. Arch. Microbiol. 2022, 204, 577. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, J.; Du, G.; Chen, J.; Ren, T.; Wang, J.; Zhao, X. The Influence of Seasons on the Composition of Microbial Communities and the Content of Lactic Acid during the Fermentation of Fen-Flavor Baijiu. Fermentation 2022, 8, 740. [Google Scholar] [CrossRef]
- Wang, S.; Li, Z.; Huang, D.; Luo, H. Contribution of microorganisms from pit mud to volatile flavor compound synthesis in fermented grains for Nongxiangxing Baijiu brewing. J. Sci. Food Agric. 2023, 104, 778–787. [Google Scholar] [CrossRef]
- Liu, S.; Ren, D.; Qin, H.; Yin, Q.; Yang, Y.; Liu, T.; Mao, J. Exploring major variable factors influencing flavor and microbial characteristics of upper jiupei. Food Res. Int. 2023, 172, 113057. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wu, M.; Zhao, D.; Zheng, J.; Dai, M.; Li, X.; Sun, B. Simulated Fermentation of Strong-Flavor Baijiu through Functional Microbial Combination to Realize the Stable Synthesis of Important Flavor Chemicals. Foods 2023, 12, 644. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Long, Y.; Li, Q.; Yang, L.; Huang, Y.; Yu, D.; Song, W.; Zhou, M.; Xu, G.; Huang, C.; et al. Propidium Monoazide Combined With RT-qPCR Detects Infectivity of Porcine Epidemic Diarrhea Virus. Front. Vet. Sci. 2022, 9, 931392. [Google Scholar] [CrossRef]
- Sun, Y.Q.; Ge, Y. Relic DNA effects on the estimates of bacterial community composition and taxa dynamics in soil. Appl. Microbiol. Biotechnol. 2023, 107, 4109–4117. [Google Scholar] [CrossRef]
- Liu, F.; Lu, H.; Dong, B.; Huang, X.; Cheng, H.; Qu, R.; Xu, Z.Z. Systematic Evaluation of the Viable Microbiome in the Human Oral and Gut Samples with Spike-in Gram+/- Bacteria. Msystems 2023, 8, e0073822. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.-C.; Li, Y.; Qiu, W.-W.; Wu, X.-Q.; Xu, B.-X.; Liang, Y.-T.; Liu, B.; Chen, S.-J.; Rao, P.-F.; Ni, L. Development of propidium monoazide combined with real-time quantitative PCR (PMA-qPCR) assays to quantify viable dominant microorganisms responsible for the traditional brewing of Hong Qu glutinous rice wine. Food Control 2016, 66, 69–78. [Google Scholar] [CrossRef]
- Xue, Y.; Abdullah Al, M.; Chen, H.; Xiao, P.; Zhang, H.; Jeppesen, E.; Yang, J. Relic DNA obscures DNA-based profiling of multiple microbial taxonomic groups in a river-reservoir ecosystem. Mol. Ecol. 2023, 32, 4940–4952. [Google Scholar] [CrossRef] [PubMed]
- Guangxun, T. Research on Composition, Source and Dynamic Change Regularity of Microorganisms Related to Chinese Strong Flavor Liquor Brewing Based on Viable Microorganisms Data. Master’s Thesis, Huazhong Agricultural University, Wuhan, China, 2020. [Google Scholar]
- Kangli, W. Study of the Diversity and Metabolic Characteristics of Microbial Community Harbored in Fermented Grains for the Chinese Strong-Flavor Baijiu Production Based on Metatranscriptome. Master’s Thesis, Zhengzhou University of Light Industry, Zhengzhou, China, 2021. [Google Scholar]
- Li, X.; Tan, G.; Chen, P.; Cai, K.; Dong, W.; Peng, N.; Zhao, S. Uncovering acid resistance genes in lactic acid bacteria and impact of non-viable bacteria on bacterial community during Chinese strong-flavor baijiu fermentation. Food Res. Int. 2023, 167, 112741. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Du, H.; Zhang, Y.; Xu, Y. Unraveling Core Functional Microbiota in Traditional Solid-State Fermentation by High-Throughput Amplicons and Metatranscriptomics Sequencing. Front. Microbiol. 2017, 8, 1294. [Google Scholar] [CrossRef]
- Dan, H.; Song, X.; Xiang, G.; Song, C.; Dai, H.; Shao, Y.; Huang, D.; Luo, H. The response pattern of the microbial community structure and metabolic profile of jiupei to Bacillus subtilis JP1 addition during baijiu fermentation. J. Sci. Food Agric. 2024. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, X.; Qiu, S.; Zeng, C.; Zhang, Y.; Cui, D.; Wu, H.; Ban, S. Microbial community and interaction between lactic acid bacteria andmicroorganisms in liquid fermented food: A review. Microbiol. China 2021, 48, 960–973. [Google Scholar] [CrossRef]
- Zeng, C.; Qiu, S.; Hu, B.; Chen, M.; Xu, J.; Wang, X. Research progress of anaerobic microorganisms in liquor brewing system. China Brew. 2015, 34, 24–27. [Google Scholar]
- Zhu, Z.Y.; Huang, Y.G. Structure and Diversity Analysis of Mold Community in Main Maotai-flavor Baijiu Brewing Areas of Maotai Town Using High-throughput Sequencing. Food Sci. 2021, 42, 150–156. [Google Scholar]
- Wu, X.; Jiang, Q.; Wang, Z.; Xu, Y.; Chen, W.; Sun, J.; Liu, Y. Diversity, enzyme production and antibacterial activity of Bacillus strains isolated from sesame-flavored liquor Daqu. Arch. Microbiol. 2021, 203, 5831–5839. [Google Scholar] [CrossRef]
- Sun, S.J.; Zhai, L.; Yu, X.J.; Xu, L.; Yao, S. Influences of Saccharomycopsis fibuligera ClCC 33077 on microbial community and functional characteristics of high temperature Daqu in sesame flavor Baijiu. Food Ferment. Ind. 2023, 49, 99–105. [Google Scholar] [CrossRef]
- Niutian, Y. Study on the Microbial Community Structure of Light-Flavor Baijiu Brewing and Its Correlation with Physicochemical Indicators and Flavor Components. Master’s Thesis, Shanxi Agricultural University, Jinzhong, China, 2022. [Google Scholar]
- Cui, S.; Dai, Y. Change Rules of Microorganisms and Fermented Grains during Fermentation of Jiangxiang Baijiu. Liquor-Mak. Sci. Technol. 2021, 6, 65–68. [Google Scholar] [CrossRef]
- Li, R.; Xu, Y.; Wang, D. Interaction mechanism of non-Saccharomyces yeast and Saccharomyces cerevisiae in mixed fermentation of Mijiu. Food Ferment. Ind. 2024, 50, 41–47. [Google Scholar] [CrossRef]
- Tang, J.; Wang, H.Y.; Xu, Y. Effect of mixed culture of Saccharomyces cerevisiae and Pichia anomala on fermentation efficiency and flavor compounds in Chinese Liquor. Microbiol. China 2012, 39, 921–930. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bo, T.; Zhang, J.; Zong, E.; Lv, N.; Bai, B.; Yang, Y.; Zhang, J.; Fan, S. Selective Elucidation of Living Microbial Communities in Fermented Grains of Chinese Baijiu: Development of a Technique Integrating Propidium Monoazide Probe Pretreatment and Amplicon Sequencing. Foods 2024, 13, 1782. https://doi.org/10.3390/foods13111782
Bo T, Zhang J, Zong E, Lv N, Bai B, Yang Y, Zhang J, Fan S. Selective Elucidation of Living Microbial Communities in Fermented Grains of Chinese Baijiu: Development of a Technique Integrating Propidium Monoazide Probe Pretreatment and Amplicon Sequencing. Foods. 2024; 13(11):1782. https://doi.org/10.3390/foods13111782
Chicago/Turabian StyleBo, Tao, Jiaojiao Zhang, Enxiang Zong, Na Lv, Baoqing Bai, Yukun Yang, Jinhua Zhang, and Sanhong Fan. 2024. "Selective Elucidation of Living Microbial Communities in Fermented Grains of Chinese Baijiu: Development of a Technique Integrating Propidium Monoazide Probe Pretreatment and Amplicon Sequencing" Foods 13, no. 11: 1782. https://doi.org/10.3390/foods13111782
APA StyleBo, T., Zhang, J., Zong, E., Lv, N., Bai, B., Yang, Y., Zhang, J., & Fan, S. (2024). Selective Elucidation of Living Microbial Communities in Fermented Grains of Chinese Baijiu: Development of a Technique Integrating Propidium Monoazide Probe Pretreatment and Amplicon Sequencing. Foods, 13(11), 1782. https://doi.org/10.3390/foods13111782