
Direct Determination of Glyphosate and Its Metabolites in Foods of Animal Origin by Liquid Chromatography–Tandem Mass Spectrometry

Abstract
1. Introduction
2. Materials and Methods
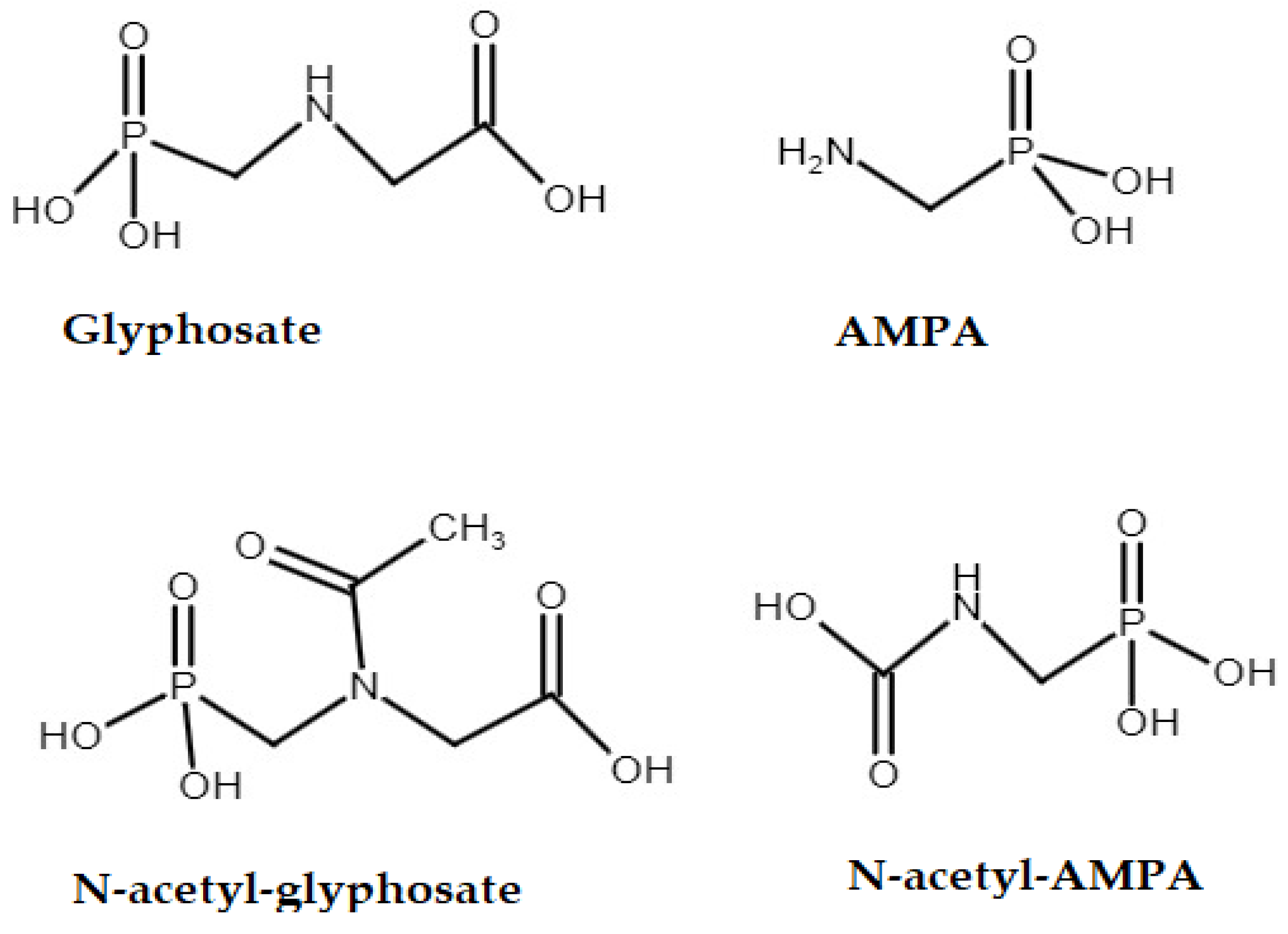
2.1. Chemicals and Materials
2.2. Standard Preparation
2.3. Instrumentation
2.4. Sample Preparation
2.5. Method Validation Procedure
2.5.1. Linearity
2.5.2. Limits of Quantification
2.5.3. Precision
2.5.4. Recovery Rate
2.5.5. Measurement Uncertainty
3. Results and Discussion
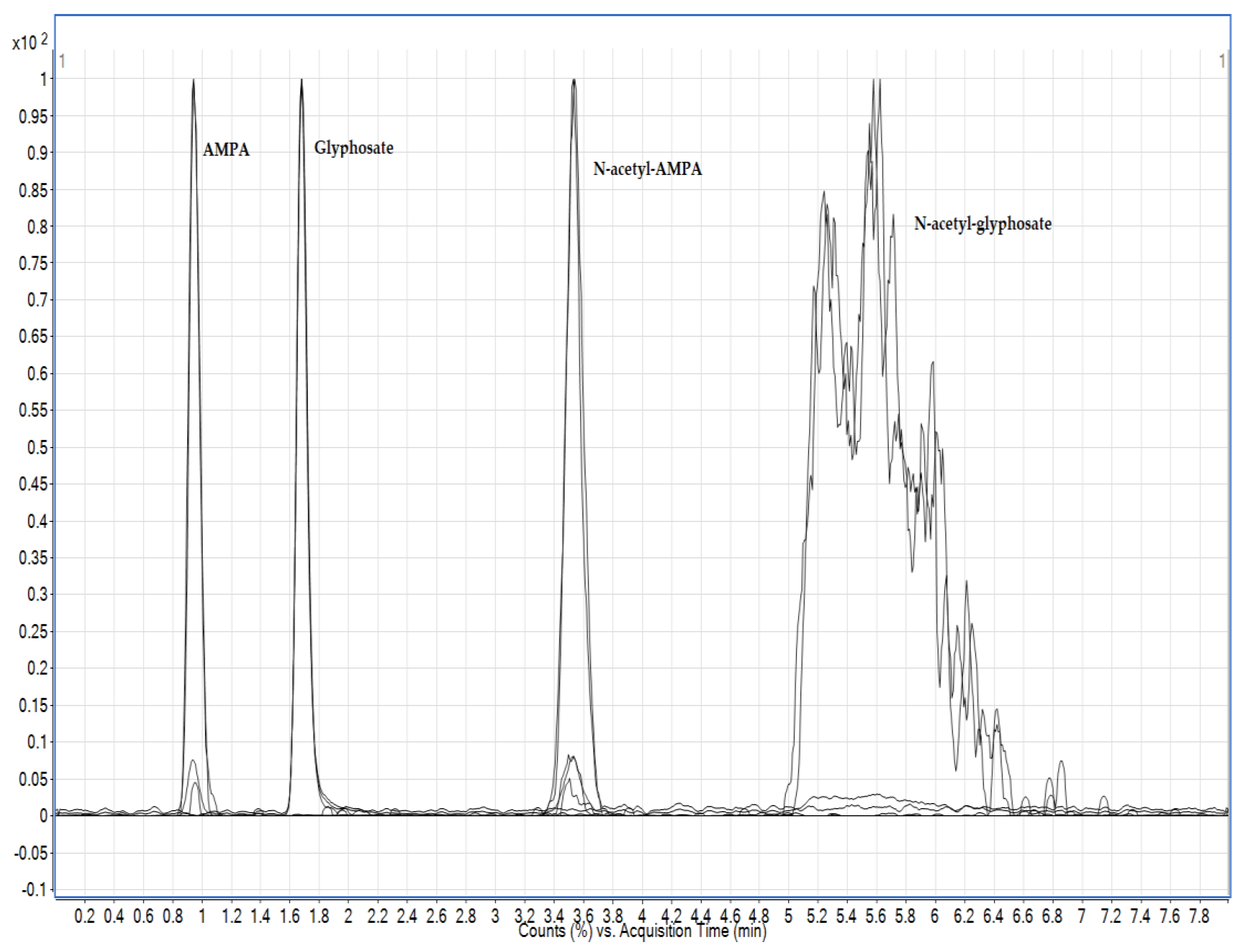
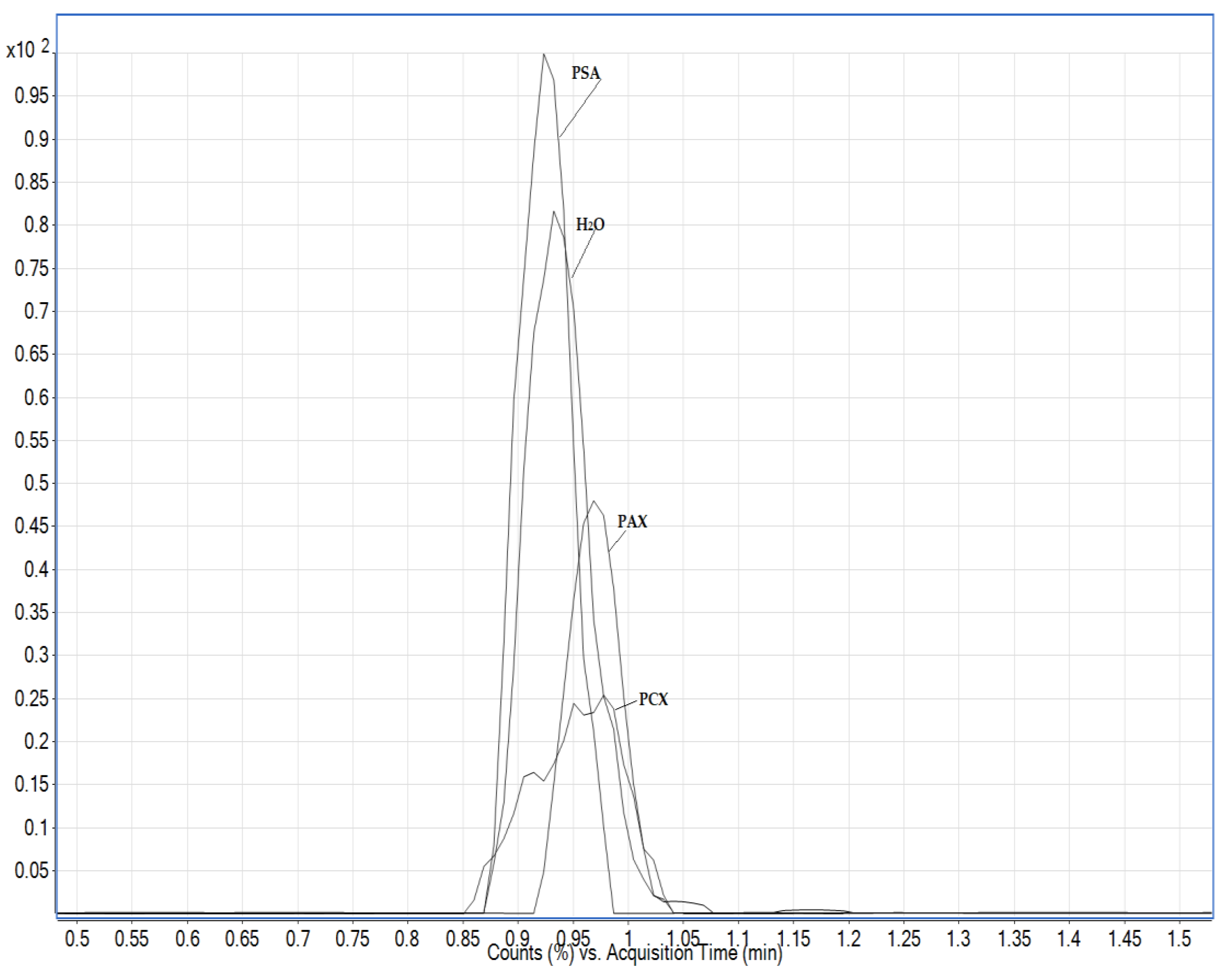
3.1. Chromatography Optimisation
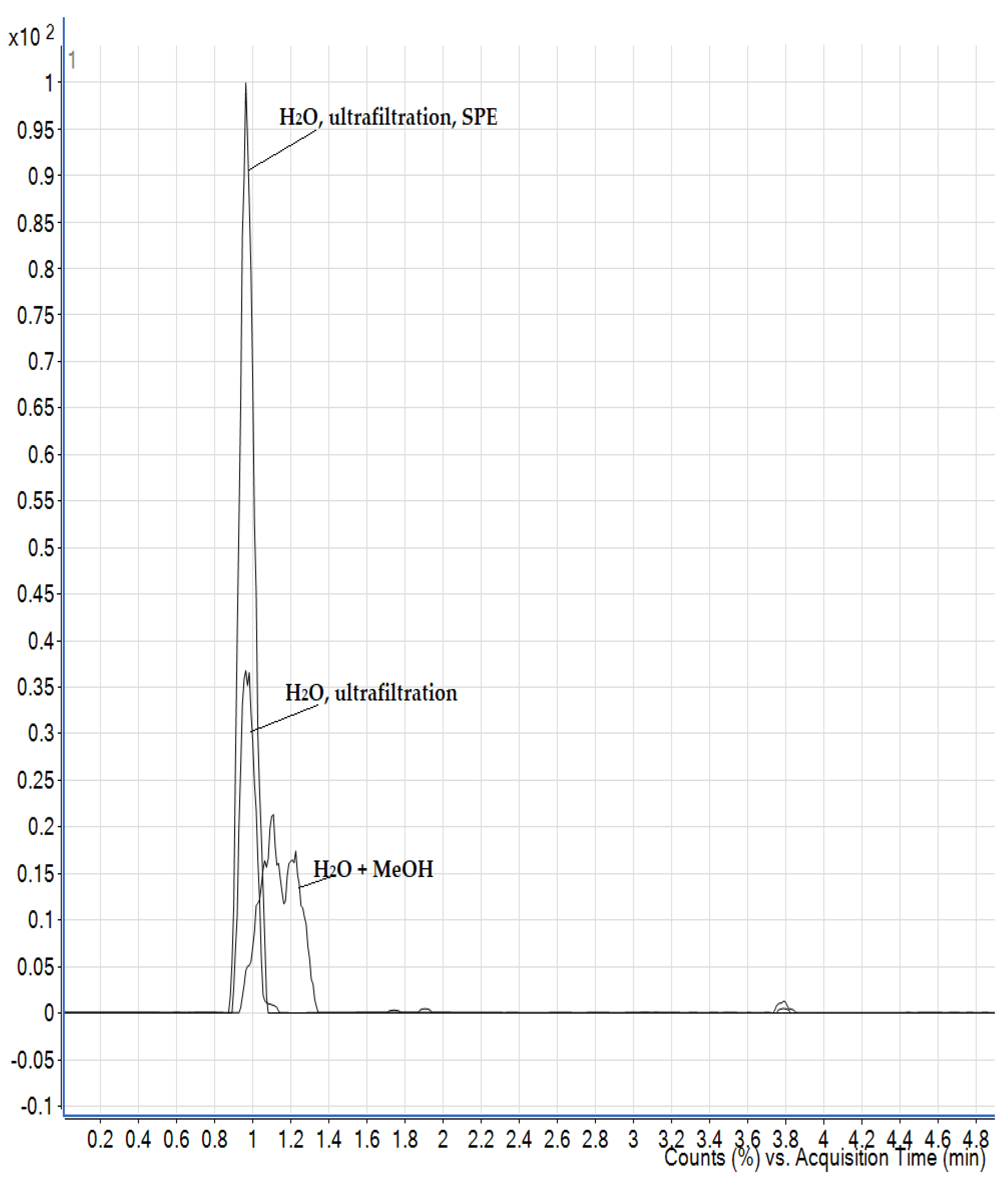
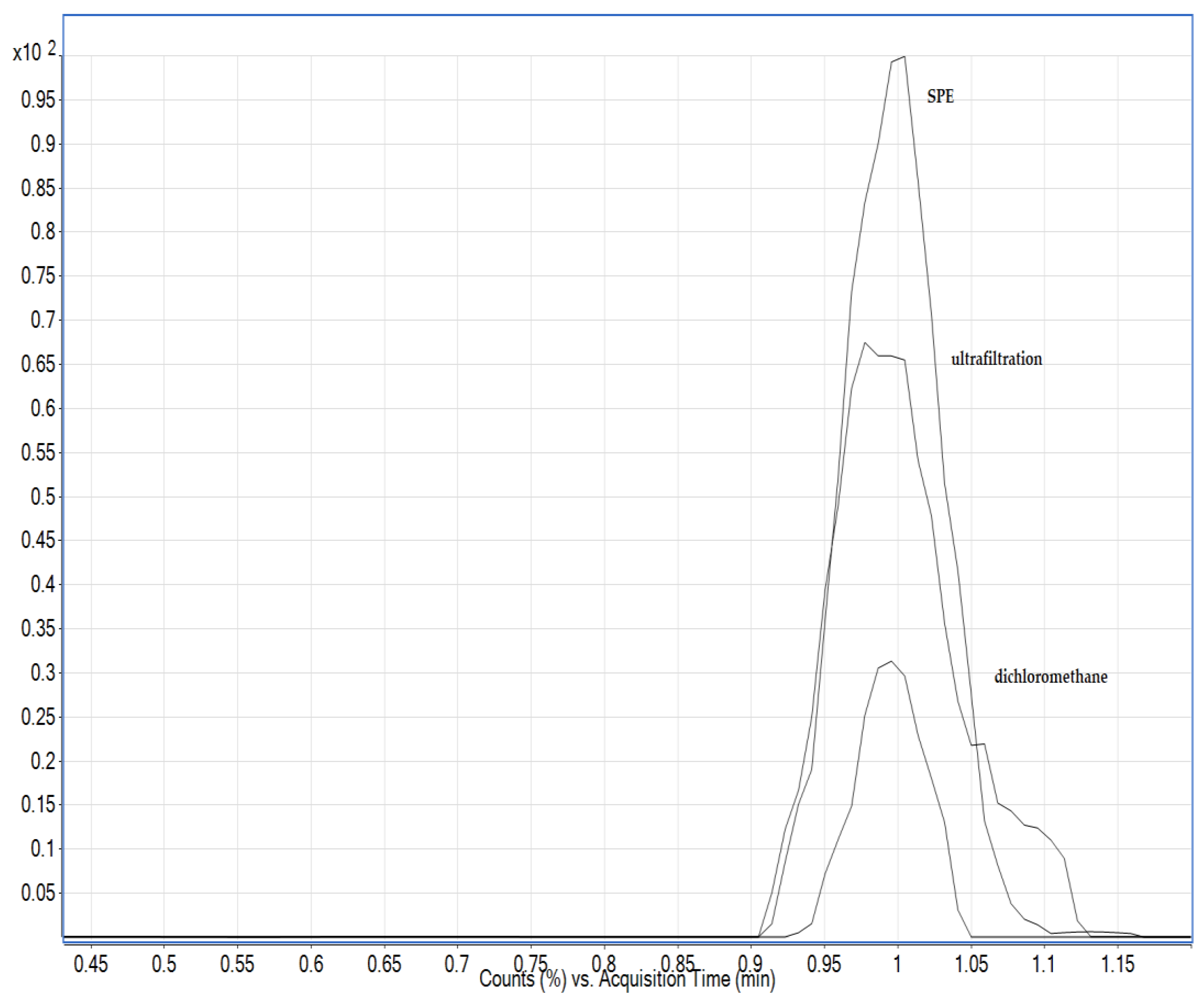
3.2. Optimisation of the Extraction Procedure
3.3. Analytical Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Food Safety Authority (EFSA). Peer review of the pesticide risk assessment of the potential endocrine activity of glyphosate. EFSA J. 2017, 15, 4979. [Google Scholar] [CrossRef]
- European Commission. Regulation (EC) No 293/2013 of 20 March 2013 amending Annexes II and III to Regulation (EC) No 396/2005 of the European Parliament and of the Council as regards maximum residue levels for emamectin benzoate, etofenprox, etoxazole, flutriafol, glyphosate, phosmet, pyraclostrobin, spinosad and spirotetramat in or on certain products. Off. J. Eur. Union 2013, L96/1, 252–281. [Google Scholar]
- European Food Safety Authority (EFSA). Review of the existing maximum residue levels for glyphosate according to Article 12 of Regulation (EC) No 396/2005–revised version to take into account omitted data. EFSA J. 2019, 17, e05862. [Google Scholar] [CrossRef]
- European Commission. Commission Implementing Regulation (EU) 2021/601 of 13 April 2021 concerning a coordinated multiannual control programme of the Union for 2022, 2023 and 2024 to ensure compliance with maximum residue levels of pesticides and to assess the consumer exposure to pesticide residues in and on food of plant and animal origin. Off. J. Eur. Union 2021, 64, 29–41. [Google Scholar]
- Nørskov, N.P.; Jensen, S.K.; Sørensen, M.T. Robust and highly sensitive micro liquid chromatography–tandem mass spectrometry method for analyses of polar pesticides (glyphosate, aminomethylphosfonic acid, N-acetyl glyphosate and N-acetyl aminomethylphosfonic acid) in multiple biological matrices. J. Chromatogr. A 2019, 1605, 360343. [Google Scholar] [CrossRef]
- Raina-Fulton, R. A Review of Methods for the Analysis of Orphan and Difficult Pesticides: Glyphosate, Glufosinate, Quaternary Ammonium and Phenoxy Acid Herbicides, and Dithiocarbamate and Phthalimide Fungicides. J. AOAC Int. 2014, 97, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Valle, A.L.; Mello, F.C.C.; Alves-Balvedi, R.P.; Rodrigues, L.P.; Goulart, L.R. Glyphosate detection: Methods, needs and challenges. Environ. Chem. Lett. 2019, 17, 291–317. [Google Scholar] [CrossRef]
- Chiarello, M.; Jimènez-Medina, M.L.; Marín Saéz, J.; Moura, S.; Garrido Frenich, A.; Romero-González, R. Fast analysis of glufosinate, glyphosate and its main metabolite, aminomethylphosphonic acid, in edible oils, by liquid chromatographycoupled with electrospray tandem mass spectrometry. Food Addit. Contam. Part A 2019, 36, 1376–1384. [Google Scholar] [CrossRef]
- Goncharova, E.N.; Statkus, M.A.; Tsizin, G.I.; Selimov, R.N. HPLC Determination of Glyphosate, Aminomethylphosphonic Acid, and Glufosinate Using a Hypercarb Porous Graphite Adsorbent. Mosc. Univ. Chem. Bull. 2018, 73, 265–271. [Google Scholar] [CrossRef]
- Verdini, E.; Lattanzio, V.M.T.; Ciasca, B.; Fioroni, L.; Pecorelli, I. Improved Method for the Detection of Highly Polar Pesticides and Their Main Metabolites in Foods of Animal Origin: Method Validation and Application to Monitoring Programme. Separations 2023, 10, 44. [Google Scholar] [CrossRef]
- Anastassiades, M.; Kolberg, D.I.; Eichhorn, E.; Wachtler, A.K.; Benkenstein, A.; Zechmann, S.; Mack, D.; Wildgrube, C.; Barth, A.; Sigalov, I.; et al. Quick Method for the Analysis of Numerous Highly Polar Pesticides in Food Involving Extraction with Acidified Methanol and LC-MS/MS Measurement. In Food of Animal Origin (QuPPe-AO-Method), Version 3.2; European Comission: Brussels, Belgium, 2019; Available online: https://www.eurl-pesticides.eu/userfiles/file/meth_QuPPe_AO_V3_2.pdf (accessed on 5 July 2023).
- Chamkasem, N. Determination of glyphosate, maleic hydrazide, fosetyl aluminum, and ethephon in grapes by liquid chromatography/tandem mass spectrometry. J. Agric. Food Chem. 2017, 65, 7535–7541. [Google Scholar] [CrossRef] [PubMed]
- Chamkasem, N.; Harmon, T. Direct determination of glyphosate, glufosinate, and AMPA in soybean and corn by liquid chromatography/tandem mass spectrometry. Anal. Bioanal. Chem. 2016, 408, 4995–5004. [Google Scholar] [CrossRef] [PubMed]
- Chamkasem, N.; Morris, C.; Harmon, T. Direct Determination of Glyphosate, Glufosinate, and AMPA in milk by Liquid chromatography/tandem mass spectrometry. J. Regul. Sci. 2015, 3, 20–26. [Google Scholar] [CrossRef]
- Baker, D.R.; Levi, M.; Capodanno, E. Highly polar pesticide multi-resiude analysis in food safety by LC-MS/MS. Available online: http://www.spectroscopynow.com/details/advertorial/14cff8e8444/Highly-polar-pesticide-multi-residue-analysis-in-food-safety-by-LC-MSMS.html (accessed on 18 January 2019).
- Lee, J.H.; Park, H.N.; Park, H.J.; Heo, S.; Park, S.S.; Park, S.-K.; Baek, S.Y. Development and Validation of LC–MS/MS and LC-Q-Orbitrap/MS Methods for Determination of Glyphosate in Vaccines. Chromatographia 2017, 80, 1741–1747. [Google Scholar] [CrossRef]
- Herrera López, S.; Scholten, J.; Kiedrowska, B.; de Kok, A. Method validation and application of a selective multiresidue analysis of highly polar pesticides in food matrices using hydrophilic interaction liquid chromatography and mass spectrometry. J. Chromatogr. A 2019, 1594, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Herrera López, S.; Dias, J.; de Kok, A. Analysis of highly polar pesticides and their main metabolites in animal origin matrices by hydrophilic interaction liquid chromatography and mass spectrometry. Food Control 2020, 115, 107289. [Google Scholar] [CrossRef]
- Hao, C.; Morse, D.; Morra, F.; Zhao, X.; Yang, P.; Nunn, B. Direct aqueous determination of glyphosate and related compounds by liquid chromatography/tandem mass spectrometry using reversed-phase and weak anion-exchange mixed-mode column. J. Chromatogr. A. 2011, 1218, 5638–5643. [Google Scholar] [CrossRef]
- Liao, Y.; Berthion, J.M.; Colet, I.; Merlo, M.; Nougadere, A.; Hu, R. Validation and application of analytical method for glyphosate and glufosinate in foods by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2018, 1549, 31–38. [Google Scholar] [CrossRef]
- Gasparini, M.; Angelone, B.; Ferretti, E. Glyphosate and other highly polar pesticides in fruit, vegetables and honey using ion chromatography coupled with high resolution mass spectrometry: Method validation and its applicability in an official laboratory. J. Mass Spectrom. 2020, 55, e4624. [Google Scholar] [CrossRef]
- Chamkasem, N.; Vargo, J. Development and Independent Laboratory Validation of an Analytical Method for the Direct Determination of Glyphosate, Glufosinate, and Aminomethylphosphonic Acid in Honey by Liquid Chromatography/Tandem Mass Spectrometry. J. Regul. Sci. 2017, 5, 1–9. [Google Scholar] [CrossRef]
- Rampazzo, G.; Gazzotti, T.; Zironi, E.; Pagliuca, G. Glyphosate and Glufosinate Residues in Honey and Other Hive Products. Foods 2023, 12, 1155. [Google Scholar] [CrossRef] [PubMed]
- Zoller, O.; Rhyn, P.; Rupp, H.; Zarn, J.A.; Geiser, C. Glyphosate residues in Swiss market foods: Monitoring and risk evaluation. Food Addit. Contam. 2018, 11, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.K.; Wujcik, C.E.; McGuire, M.K.; McGuire, M.A. Validation of reliable and selective methods for direct determination of glyphosate and aminomethylphosphonic acid in milk and urine using LC-MS/MS. J. Environ. Sci. Health B 2016, 51, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Rubio, F.; Guo, E.; Kamp, L. Survey of glyphosate residues in honey, corn and soy products. J. Environ. Anal. Toxicol. 2014, 5, 1000249. [Google Scholar] [CrossRef]
- Chiesa, L.M.; Nobile, M.; Panseri, S.; Arioli, F. Detection of glyphosate and its metabolites in food of animal origin based on ion-chromatographyhigh resolution mass spectrometry (IC-HRMS). Food Addit. Contam. A 2019, 36, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Verdini, E.; Pecorelli, I. The Current Status of Analytical Methods Applied to the Determination of Polar Pesticides in Food of Animal Origin: A Brief Review. Foods 2022, 11, 1527. [Google Scholar] [CrossRef] [PubMed]
- Berg, C.J.; King, H.P.; Delenstarr, G.; Kumar, R.; Rubio, F.; Glaze, T. Glyphosate residue concentrations in honey attributed through geospatial analysis to proximity of large-scale agriculture and transfer off-site by bees. PLoS ONE 2018, 13, e0198876. [Google Scholar] [CrossRef] [PubMed]
- SANTE/11312/2021: Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed Implemented by 1 January 2022. Available online: https://www.eurl-pesticides.eu/userfiles/file/EurlALL/SANTE_11312_2021.pdf (accessed on 8 November 2023).
- Núňez, O.; Lucci, P. Applications and uses of formic acid in liquid chromatography-mass spectrometry analysis. In Advances in Chemical Research; Nova Science Publishers: Hauppauge, NY, USA, 2014; pp. 71–86. Available online: https://www.researchgate.net/publication/267508772_Applications_and_Uses_of_Formic_Acid_in_Liquid_Chromatography-Mass_Spectrometry_Analysis (accessed on 10 November 2023).
- Pareja, L.; Jesús, F.; Heinzen, H.; Hernando, M.D.; Rajski, Ł.; Fernández-Alba, A.R. Evaluation of glyphosate and AMPA in honey by water extraction followed by ion chromatography mass spectrometry. A pilot monitoring study. Anal. Methods 2019, 11, 2123–2128. [Google Scholar] [CrossRef]
- Thompson, T.S.; van den Heever, J.P.; Limanowka, R.E. Determination of glyphosate, AMPA, and glufosinate in honey by onlinesolid-phase extraction-liquid chromatography-tandem mass spectrometry. Food Addit. Contam. Part A 2019, 36, 434–446. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix | Animal Species | Existing MRL (mg kg−1) | Proposed MRL According to EFSA (mg kg−1) |
|---|---|---|---|
| Muscle | Pig, cattle, sheep, goat, horse, poultry | 0.05 | 0.2 |
| Fat tissue | Pig, cattle, horse, poultry | 0.05 | 0.2 |
| sheep, goat | 0.05 | 0.3 | |
| Liver | Pig | 0.05 | 0.4 |
| Cattle | 0.2 | 0.7 | |
| Sheep, goat | 0.05 | 0.9 | |
| Horse | 0.05 | 0.7 | |
| Poultry | 0.05 | 0.2 | |
| Kidney | Pig | 0.5 | 3 |
| Cattle | 2 | 7 | |
| Sheep, goat | 0.05 | 10 | |
| Horse | 0.05 | 7 | |
| Milk | Sheep, goat, cattle, Horse | 0.05 | 0.1 |
| Eggs | Birds | 0.05 | 0.1 |
| Honey | 0.05 | No recommendation |
| Compound | ISTD | Ion Precursor | Ion Product (m/z) | Fragment (V) | Collision Energy (V) |
|---|---|---|---|---|---|
| AMPA | AMPA-13C2, 15N | 110 | 79 63 | 70 | 28 18 |
| Glyphosate | Glyphosate-13C2, 15N | 170 | 88.2 60.2 | 50 | 2 12 |
| N-acetyl-glyphosate | N-acetyl-glyphosate-D3 | 210 | 124 150 | 70 | 12 6 |
| N-acetyl-AMPA | - | 152 | 134 110 63 | 70 | 8 8 26 |
| Glyphosate-13C2, 15N | - | 173 | 91 62.2 | 50 | 2 12 |
| AMPA-13C2, 15N | - | 112 | 79 63 | 80 | 30 16 |
| N-acetyl-glyphosate-D3 | - | 213 | 153 126 | 70 | 6 12 |
| Matrix | Analyte | AMPA | Glyphosate | N-acetyl-AMPA | N-acetyl-glyphosate |
|---|---|---|---|---|---|
| Fat tissue | r2 | 0.985 | 0.996 | 0.992 | 0.991 |
| Linearity/ng mL−1 | 12.5–100 | 12.5–100 | 12.5–100 | 12.5–100 | |
| Liver | r2 | 0.967 | 0.996 | 0.992 | 0.993 |
| Linearity/ng mL−1 | 10–100 | 5–100 | 5–100 | 5–100 | |
| Eggs | r2 | 0.978 | 0.996 | 0.988 | 0.987 |
| Linearity/ng mL−1 | 2.5–60 | 4–60 | 4–60 | 2.5–60 | |
| Milk | r2 | 0.984 | 0.983 | 0.961 | 0.986 |
| Linearity/ng mL−1 | 20–160 | 5–160 | 20–160 | 20–160 | |
| Honey | r2 | 0.992 | 0.993 | 0.996 | 0.995 |
| Linearity/ng mL−1 | 5–160 | 5–160 | 5–160 | 5–160 |
| Limit of Quantification (mg kg−1) in Different Matrixes | |||||
|---|---|---|---|---|---|
| Analyte | Fat Tissue | Liver | Eggs | Milk | Honey |
| AMPA | 0.025 | 0.2 | 0.04 | 0.2 | 0.1 |
| Glyphosate | 0.025 | 0.05 | 0.04 | 0.025 | 0.05 |
| N-acetyl-AMPA | 0.025 | 0.1 | 0.04 | 0.1 | 0.05 |
| N-acetyl-glyphosate | 0.025 | 0.2 | 0.04 | 0.04 | 0.08 |
| Expended Measurement Uncertainty (%) in Different Matrixes | |||||
|---|---|---|---|---|---|
| Analytes | Fat Tissue | Liver | Eggs | Milk | Honey |
| AMPA | 15.7 | 41.9 | 28.6 | 37.6 | 32.9 |
| Glyphosate | 10.8 | 13.4 | 12.4 | 13.9 | 31.7 |
| N-acetyl AMPA | 13.9 | 21.0 | 21.2 | 20.5 | 15.7 |
| N-acetyl glyphosate | 26.0 | 12.8 | 25.1 | 22.0 | 22.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denžić Lugomer, M.; Bilandžić, N.; Pavliček, D.; Novosel, T. Direct Determination of Glyphosate and Its Metabolites in Foods of Animal Origin by Liquid Chromatography–Tandem Mass Spectrometry. Foods 2024, 13, 2451. https://doi.org/10.3390/foods13152451
Denžić Lugomer M, Bilandžić N, Pavliček D, Novosel T. Direct Determination of Glyphosate and Its Metabolites in Foods of Animal Origin by Liquid Chromatography–Tandem Mass Spectrometry. Foods. 2024; 13(15):2451. https://doi.org/10.3390/foods13152451
Chicago/Turabian StyleDenžić Lugomer, Marija, Nina Bilandžić, Damir Pavliček, and Tiana Novosel. 2024. "Direct Determination of Glyphosate and Its Metabolites in Foods of Animal Origin by Liquid Chromatography–Tandem Mass Spectrometry" Foods 13, no. 15: 2451. https://doi.org/10.3390/foods13152451
APA StyleDenžić Lugomer, M., Bilandžić, N., Pavliček, D., & Novosel, T. (2024). Direct Determination of Glyphosate and Its Metabolites in Foods of Animal Origin by Liquid Chromatography–Tandem Mass Spectrometry. Foods, 13(15), 2451. https://doi.org/10.3390/foods13152451