
Study on the Inhibitory Effect of Bioactive Peptides Derived from Yak Milk Cheese on Cholesterol Esterase
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Instruments and Equipment
2.3. Experimental Methods
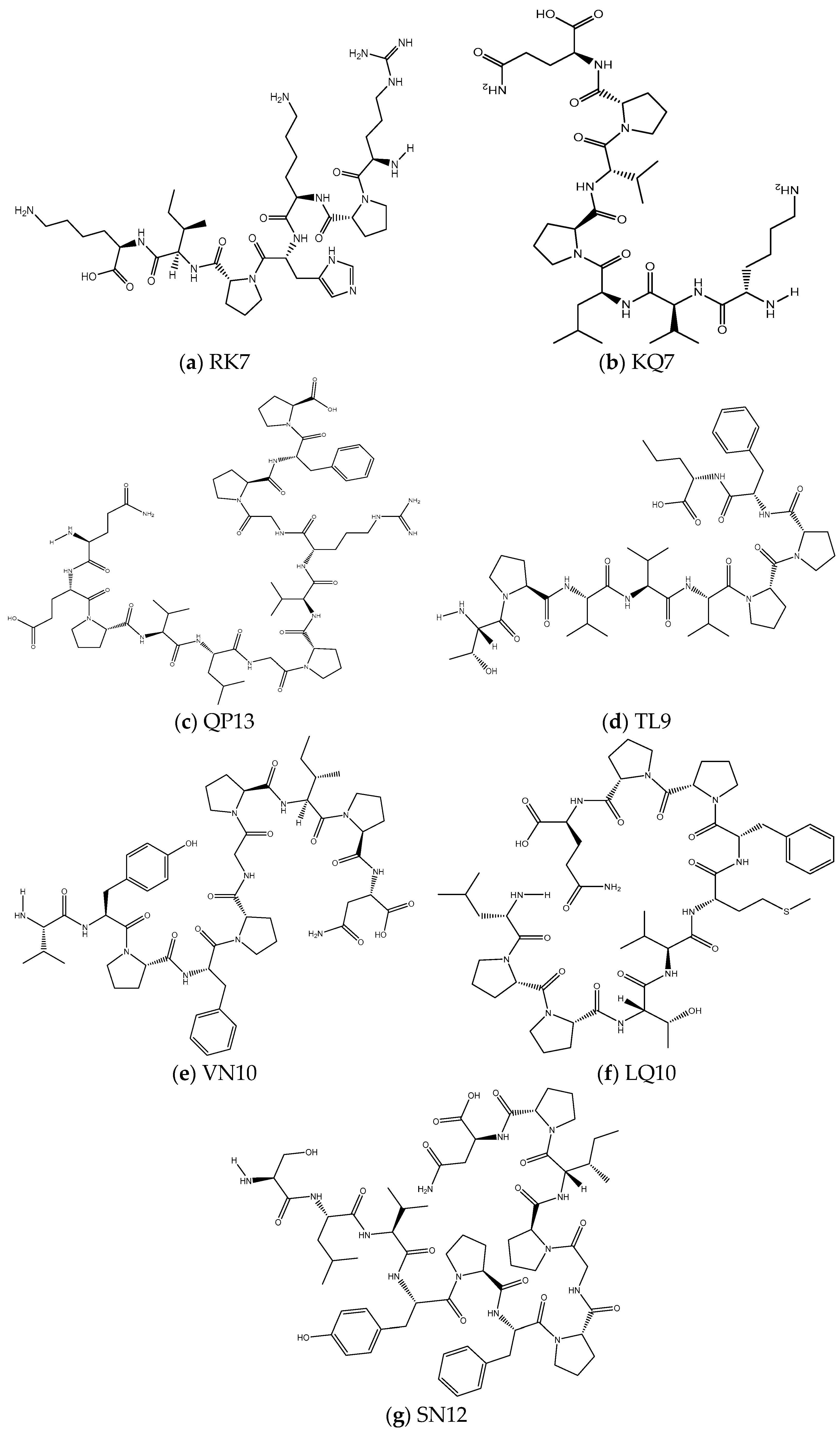
2.3.1. Prediction of Physicochemical Properties of Peptides RK7, KQ7, QP13, VN10, TL9, LQ10, and SN12
2.3.2. Structure Optimization and Processing of Peptides and Cholesterol Esterase
2.3.3. Molecular Docking
2.3.4. GROMACS Molecular Dynamics Simulation
2.3.5. Synthesis and CE Inhibitory Activity Assay of Peptides RK7, KQ7, QP13, and VN10
2.3.6. Simulating the Impact of Gastrointestinal Digestion on α-Amylase Inhibitory Activity
2.3.7. Data Processing
3. Results and Discussion
3.1. Physicochemical Characterisation of RK7, KQ7 QP13, TL9, VN10, SN12, and LQ10 Peptides
3.2. Molecular Docking Using LibDock
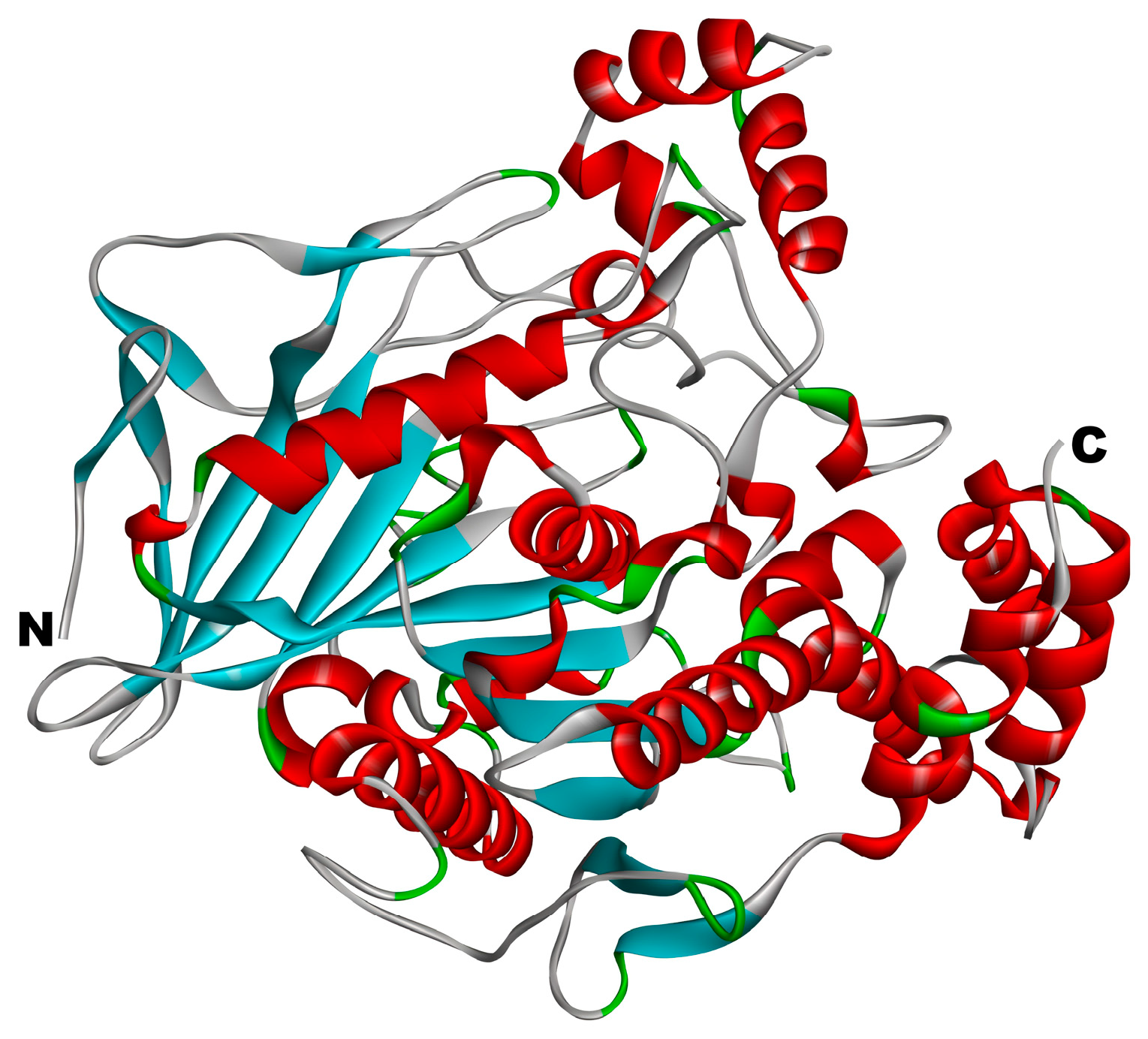
3.2.1. Reliability Verification of Molecular Docking Methods
3.2.2. Molecular Docking of CE Inhibitory Peptides
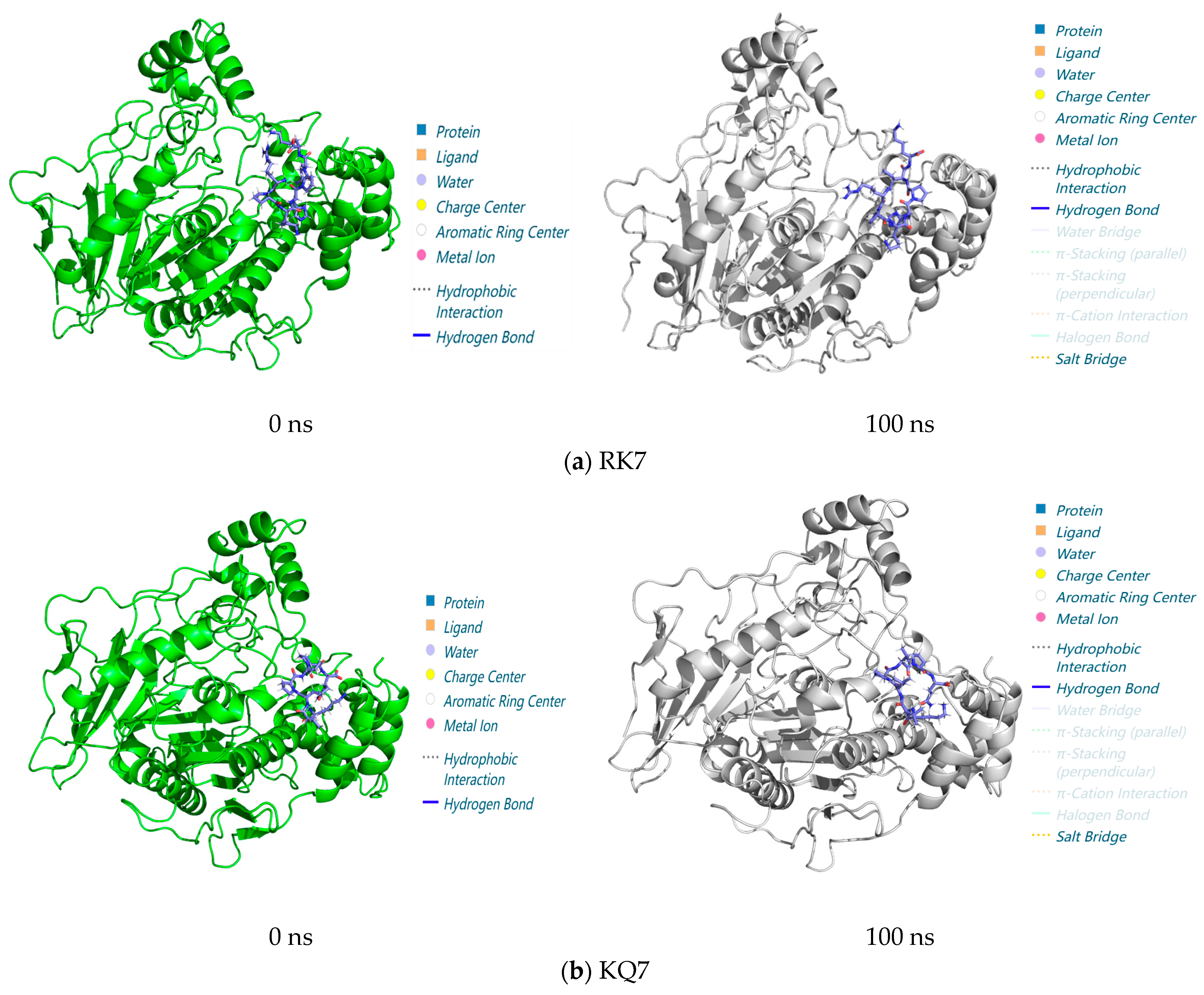
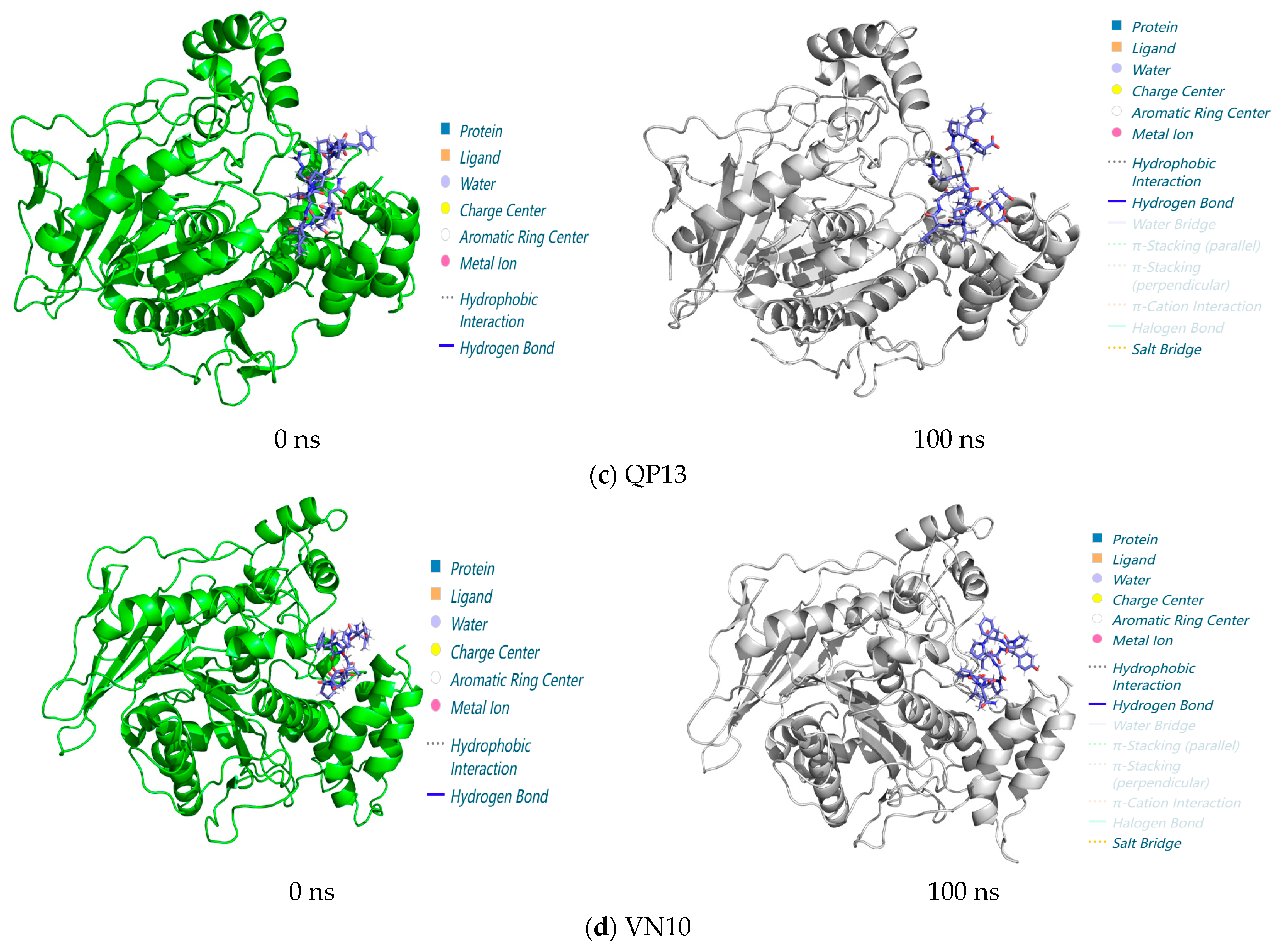
3.3. GROMACS Analysis of the Molecular Dynamics Stability of Peptide–CE Complexes
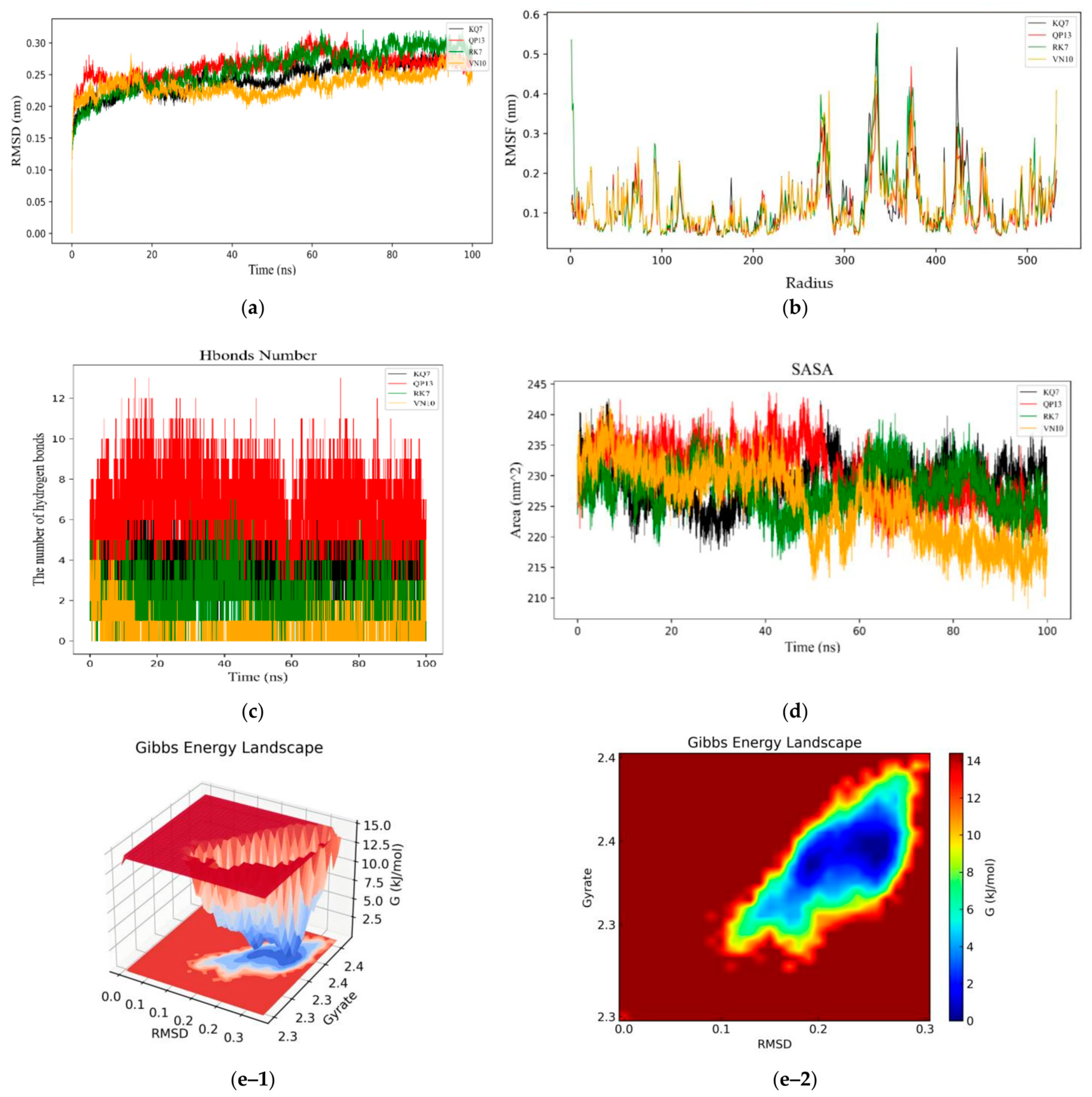
3.3.1. RMSD, RMSF, Hydrogen Bonds, SASA, and Gibbs Free Energy Stability Analysis
3.3.2. MM/GBSA Calculations and Analyses
3.3.3. Analysis of Interactions in the 0–100 ns Process of Molecular Dynamics
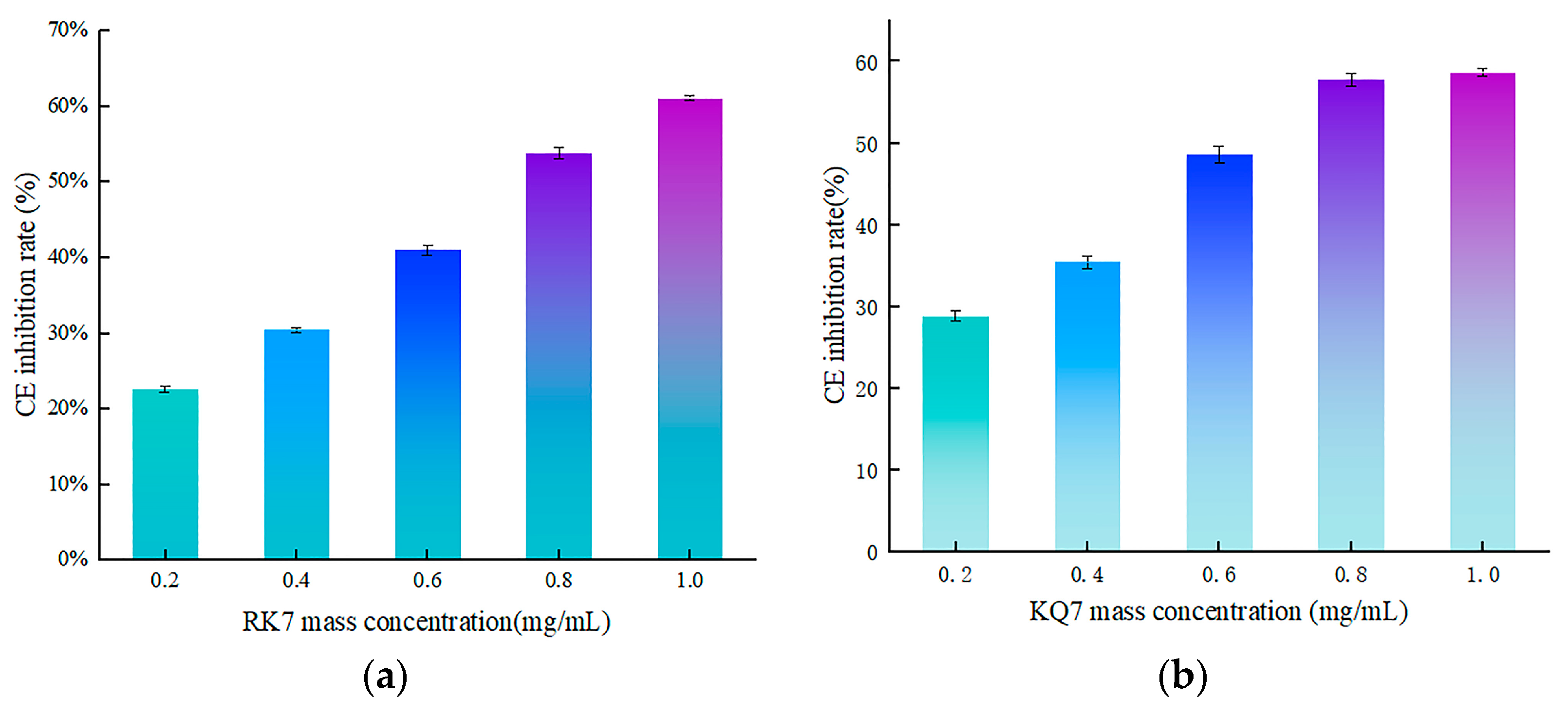
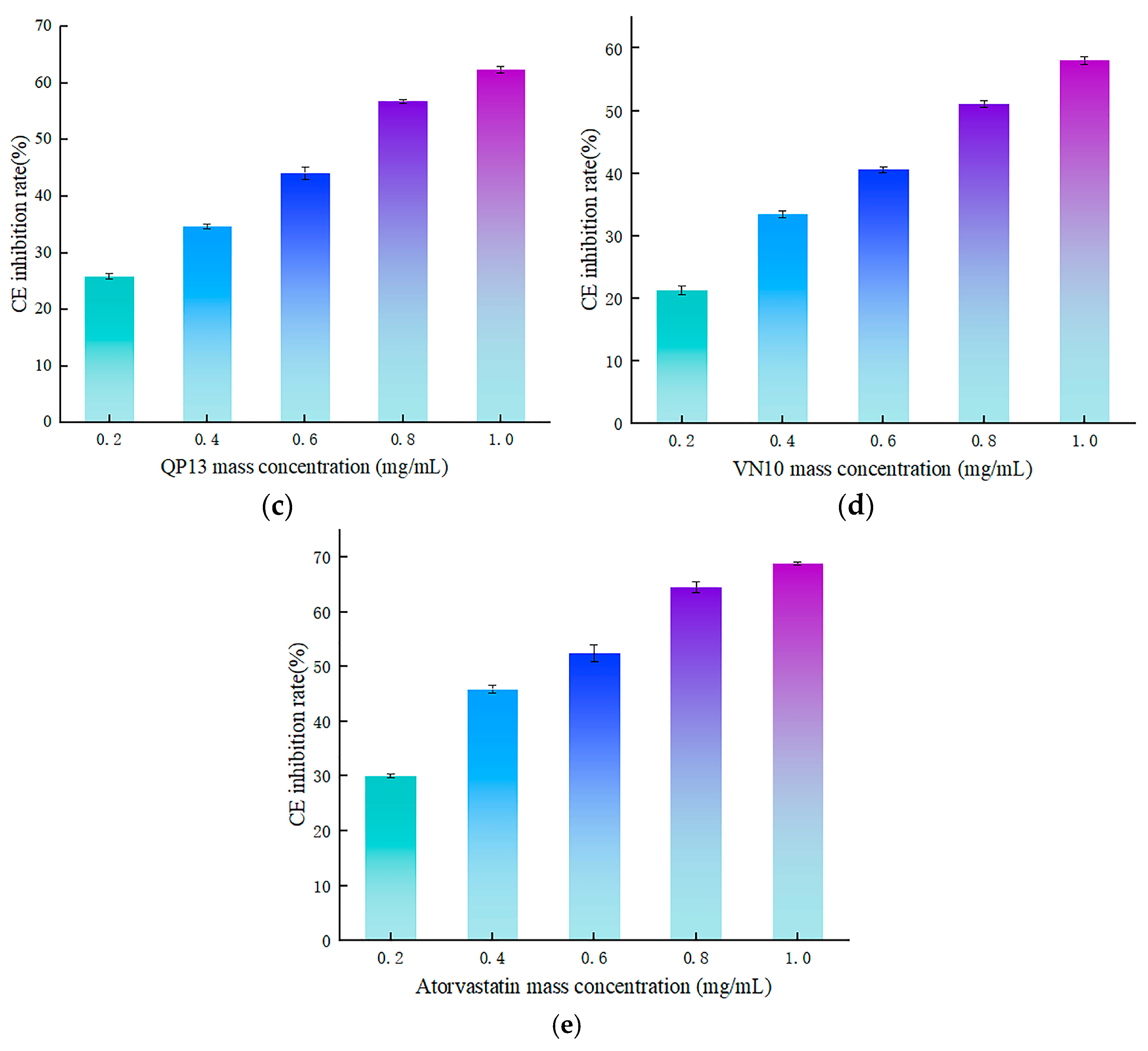
3.4. Synthesis and Validation of CE Inhibitory Activity of Peptides RK7, KQ7, QP13 and VN10
3.4.1. Synthesis of Peptides RK7, KQ7, QP13 and VN10
3.4.2. Validation of In Vitro Activity of Peptides RK7, KQ7, QP13, and VN10
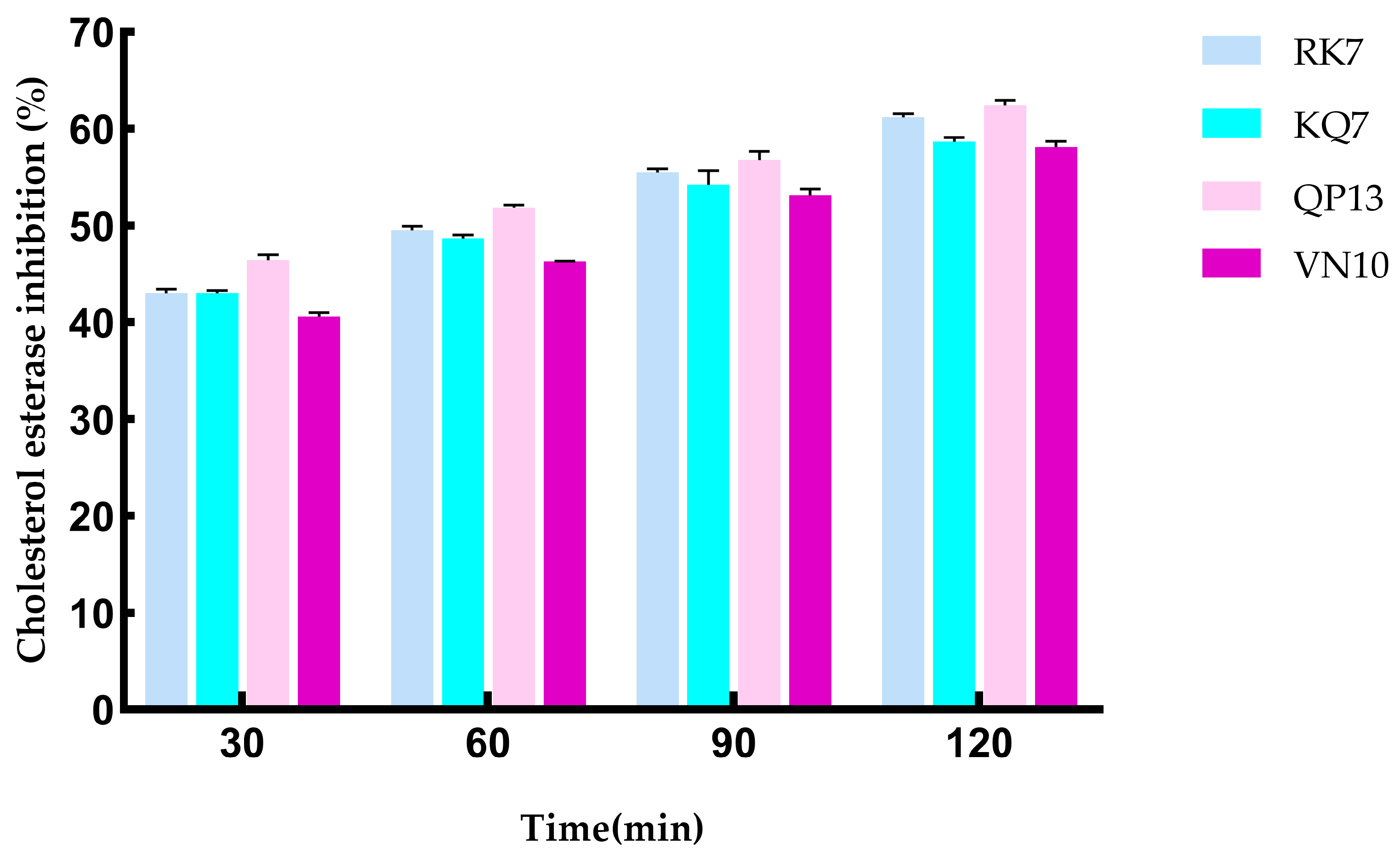
3.4.3. Simulating the Effect of Gastrointestinal Digestion on CE Inhibition Activity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rodríguez-Sánchez, I.; Ortolá, R.; Graciani, A.; Martínez-Gómez, D.; Banegas, J.R.; Rodríguez-Artalejo, F.; García-Esquinas, E. Pain Characteristics, Cardiovascular Risk Factors, and CardiovascularDisease. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2022, 77, 204–213. [Google Scholar] [CrossRef]
- Ohlsson, L. Dairy Products and Plasma Cholesterol Levels. Food Nutr. Res. 2010, 54, 5124. [Google Scholar] [CrossRef]
- Myers-Payne, S.C.; Hui, D.Y.; Brockman, H.L.; Schroeder, F. CE: A Cholesterol Transfer Protein. Biochemistry 1995, 34, 3942–3947. [Google Scholar] [CrossRef]
- Last, A.R.; Ference, J.D.; Menzel, E.R. Hyperlipidemia: Drugs for Cardiovascular Risk Reduction in Adults. Am. Fam. Physician 2017, 95, 78–87. [Google Scholar]
- Dhar, H.; Verma, S.; Dogra, S.; Katoch, S.; Vij, R.; Singh, G.; Sharma, M. Functional Attributes of Bioactive Peptides of Bovine Milk Origin and Application of In Silico Approaches for Peptide Prediction and Functional Annotations. Crit. Rev. Food Sci. Nutr. 2023, 1–23. [Google Scholar] [CrossRef]
- Yang, B.; Liang, Q. Research Progress on the Structure—Activity Relationship of Bioactive Peptides from Cheese Sources. Food Ferment. Ind. 2021, 47, 288–293. [Google Scholar] [CrossRef]
- Verruck, S.; Dantas, A.; Schwinden Prudencio, E. Functionality of the Components from Goat’s Milk, Recent Advances for Functional Dairy Products Development and Its Implications on Human Health. J. Funct. Foods 2019, 52, 243–257. [Google Scholar] [CrossRef]
- Li, M.; Liang, Q.; Song, X. Investigating the α-Amylase Inhibitory Activity of Bitter Taste Peptides RK7 and KQ7 in Yak milk cheese through Molecular Docking Technology. Food Sci. 2023, 44, 132–138. [Google Scholar] [CrossRef]
- Yang, B.; Liang, Q.; Song, X. Research on the Antibacterial Activity Difference of Bitter Taste Peptides in Hard Yak Cheese Based on Computer Virtual Technology. J. Food Biotechnol. 2021, 40, 75–87. [Google Scholar] [CrossRef]
- Yang, B.; Liang, Q.; Song, X. Antioxidant Activity and Mechanism of Peptide RK7 from Yak milk cheese Hard Cheese. J. Chin. Inst. Food Sci. Technol. 2022, 22, 40–50. [Google Scholar] [CrossRef]
- Yang, B.; Liang, Q.; Song, X. Molecular Mechanisms Characterizing the ACE Inhibitory Activity of Yak Cheese Peptides. J. Chin. Food Sci. 2022, 22, 8–17. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, Z.; Wang, H.; Zhang, F.; Guo, S.; Shen, Q. Comparison of the generation of α-glucosidase inhibitory peptides derived from prolamins of raw and cooked foxtail millet: In vitro activity, de novo sequencing, and in silico docking. Food Chem. 2023, 411, 135378. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef]
- Song, X.; Zhang, Y.; Yang, M.; Liang, Q. Isolation and characterisation of peptides from yak milk cheese hard cheese. Food Sci. 2016, 37, 160–164. [Google Scholar] [CrossRef]
- Soleymanzadeh, N.; Mirdamadi, S.; Mirzaei, M.; Kianirad, M. Novel β-casein derived antioxidant and ACE–inhibitory active peptide from camel milk fermented by Leuconostoc lactis PTCC1899: Identification and molecular docking. Int. Dairy. J. 2019, 97, 201–208. [Google Scholar] [CrossRef]
- Gurung, D.; Danielson, J.A.; Tasnim, A.; Zhang, J.T.; Zou, Y.; Liu, J.Y. Proline Isomerization: From the Chemistry and Biology to Therapeutic Opportunities. Biology 2023, 12, 1008. [Google Scholar] [CrossRef]
- López-Martínez, C.; Flores-Morales, P.; Cruz, M.; González, T.; Feliz, M.; Diez, A.; Campanera, J.M. Proline cis-trans isomerization and its implications for the dimerization of analogues of cyclopeptide stylostatin 1: A combined computational and experimental study. Phys. Chem. Chem. Phys. 2016, 18, 12755–12767. [Google Scholar] [CrossRef]
- Jiang, F.; Liu, J.B.; Ma, S.T.; Yang, M.; Chen, Y.; Liu, B.; Zhang, T. In Vitro Simulated Gastrointestinal Digestion Products of Albumin and Their Antioxidant Activities and Structural Characterization. J. Chin. Food Sci. Technol. 2021, 21, 10–18. [Google Scholar] [CrossRef]
- Karakkadparambil Sankaran, S.; Nair, A.S. Molecular dynamics and docking studies on potentially active natural phytochemicals for targeting SARS-CoV-2 main protease. J. Biomol. Struct. Dyn. 2023, 41, 6459–6475. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Esfandi, R.; Walters, M.E.; Tsopmo, A. Antioxidant properties and potential mechanisms of hydrolyzed proteins and peptides from cereals. Heliyon 2019, 5, e01538. [Google Scholar] [CrossRef]
- Ngoh, Y.Y.; Choi, S.; Gan, C.Y. The potential roles of Pinto bean (Phaseolus vulgaris cv. Pinto) bioactive peptides in regulating physiological functions: Protease activating, lipase inhibiting and bile acid binding activities. J. Funct. Foods 2017, 33, 67–75. [Google Scholar] [CrossRef]
- Nagaoka, S.; Futamura, Y.; Miwa, K.; Awano, T.; Yamauchi, K.; Kanamaru, Y.; Tadashi, K.; Kuwata, T. Identification of novel hypocholesterolemic peptides derived from bovine milk β-lactoglobulin. Biochem. Biophys. Res. Commun. 2001, 281, 11–17. [Google Scholar] [CrossRef]
- González-Ortega, O.; López-Limón, A.R.; Morales-Domínguez, J.F.; Soria-Guerra, R.E. Production and purification of recombinant hypocholesterolemic peptides. Biotechnol. Lett. 2015, 37, 41–54. [Google Scholar] [CrossRef]
- Khiari, Z.; Ndagijimana, M.; Betti, M. Low molecular weight bioactive peptides derived from the enzymatic hydrolysis of collagen after isoelectric solubilization/precipitation process of turkey by–products. Poult. Sci. 2014, 93, 2347–2362. [Google Scholar] [CrossRef]
- Pecorari, D.; Mazzanti, A.; Mancinelli, M. Atropostatin: Design and Total Synthesis of an Atropisomeric Lactone–Atorvastatin Prodrug. Molecules 2023, 28, 3176. [Google Scholar] [CrossRef]
- Kwon, D.Y.; Oh, S.W.; Yang, H.J.; Lee, S.H.; Lee, J.H.; Lee, Y.B.; Sohn, H.S.; Lee, J.S. Amino Acid Substitution of Hypocholesterolemic Peptide Originated from Glycinin Hydrolyzate. Food Sci. Biotechnol. 2002, 11, 55–61. [Google Scholar]
- Liu, F.-F.; Wang, T.; Dong, X.-Y.; Sun, Y. Rational design of affinity peptide ligand by flexible docking simulation. J. Chromatogr. A 2007, 1146, 41–50. [Google Scholar] [CrossRef]
- Ghosh, D.; Wawrzak, Z.; Pletnev, V.Z.; Li, N.; Kaiser, R.; Pangborn, W.; Jörnvall, H.; Erman, M.; Duax, W.L. Structure of uncomplexed and linoleate–bound Candida cylindracea CE. Structure 1995, 3, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Feaster, S.R.; Quinn, D.M.; Barnett, B.L. Molecular modeling of the structures of human and rat pancreatic CEs. Protein Sci. 1997, 6, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, C.S.; Tang, J.; Dyda, F.; Zhang, X.C. The crystal structure of bovine bile salt activated lipase: Insights into the bile salt activation mechanism. Structure 1997, 5, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Sivashanmugam, T.; Muthukrishnan, S.; Umamaheswari, M.; Asokkumar, K.; Subhadradevi, V.; Jagannath, P.; Madeswaran, A. Discovery of potential CE inhibitors using in silico docking studies. Bangladesh J. Pharmacol. 2013, 8, 223–229. [Google Scholar] [CrossRef]
- Neveu, E.; Popov, P.; Hoffmann, A.; Migliosi, A.; Besseron, X.; Danoy, G.; Bouvry, P.; Grudinin, S. Rapid RMSD: Rapid determination of RMSDs corresponding to motions of flexible molecules. Bioinformatics 2018, 34, 2757–2765. [Google Scholar] [CrossRef]
- Sabe, V.T.; Tolufashe, G.F.; Ibeji, C.U.; Maseko, S.B.; Govender, T.; Maguire, G.E.; Lamichhane, G.; Honarparvar, B.; Kruger, H.G. Identification of potent L, D-transpeptidase 5 inhibitors for Mycobacterium tuberculosis as potential anti-TB leads: Virtual screening and molecular dynamics simulations. J. Mol. Model. 2019, 25, 328. [Google Scholar] [CrossRef] [PubMed]
- Arshia, A.H.; Shadravan, S.; Solhjoo, A.; Sakhteman, A.; Sami, A. De novo design of novel protease inhibitor candidates in the treatment of SARS-CoV-2 using deep learning, docking, and molecular dynamic simulations. Comput. Biol. Med. 2021, 139, 104967. [Google Scholar] [CrossRef]
- Santos, L.H.S.; Ferreira, R.S.; Caffarena, E.R. Integrating Molecular Docking and Molecular Dynamics Simulations. Methods Mol. Biol. 2019, 2053, 13–34. [Google Scholar] [CrossRef]
- Mascoli, V.; Liguori, N.; Cupellini, L.; Elias, E.; Mennucci, B.; Croce, R. Uncovering the interactions driving carotenoid binding in light–harvesting complexes. Chem. Sci. 2021, 12, 5113–5122. [Google Scholar] [CrossRef]
- Ragunathan, A.; Malathi, K.; Anbarasu, A. MurB as a target in an alternative approach to tackle the Vibrio cholerae resistance using molecular docking and simulation study. J. Cell Biochem. 2018, 119, 1726–1732. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Zhong, J.; Wang, Y. A mutant T1 lipase homology modeling, and its molecular docking and molecular dynamics simulation with fatty acids. J. Biotechnol. 2021, 337, 24–34. [Google Scholar] [CrossRef]
- Mazola, Y.; Guirola, O.; Palomares, S.; Chinea, G.; Menéndez, C.; Hernández, L.; Musacchio, A. A comparative molecular dynamics study of thermophilic and mesophilic β-fructosidase enzymes. Mol. Model. 2015, 21, 228. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert. Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Sun, H.; Wang, J.; Zhu, F.; Liu, H.; Wang, Z.; Lei, T.; Li, Y.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 8. Predicting binding free energies and poses of protein–RNA complexes. RNA 2018, 24, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Luo, H.Q.; Shen, J.; Chen, C.P.; Ma, X.; Lin, C.; Ouyang, Q.; Xuan, C.X.; Liu, J.; Sun, H.B.; Liu, J. Lipid-lowering effects of oleanolic acid in hyperlipidemic patients. Chin. J. Nat. Med. 2018, 16, 339–346. [Google Scholar] [CrossRef]
- Xie, Z.; He, M.; Zhai, Y.; Xin, F.; Yu, S.; Yu, S.; Xiao, H.; Song, Y. Inhibitory kinetics and mechanism of oleanolic acid on α-glucosidase. J. Food Meas. Charact. 2021, 15, 3408–3418. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Sequence | Molecular Weight/(Da) | Isoelectric Point | Net Charge | Hydrophobic Amino Acids | Proportion of Hydrophobic Amino Acids |
|---|---|---|---|---|---|
| RK7 | 874.90 | 11.57 | 3 | P, I | 42.86% |
| KQ7 | 779.50 | 9.84 | 1 | V, L, P | 71.42% |
| QP13 | 1392.60 | 6.58 | 0 | P, V, L, G, F | 61.54% |
| TL9 | 968.19 | 3.32 | 0 | P, V, F, L | 88.89% |
| VN10 | 1100.26 | 3.63 | 0 | V, P, F, G, I | 70.00% |
| LQ10 | 1300.50 | 4.34 | 0 | L, P, V, M, F | 66.67% |
| SN12 | 1126.37 | 3.70 | 0 | L, V, P, F, G, I | 80.00% |
| RK7 | KQ7 | QP13 | VN10 | TL9 | LQ10 | SN12 | |
|---|---|---|---|---|---|---|---|
| Hydrogen bonds | Thr68 Ala67 Gln66 Asn118 Ala117 Asn122 Arg423 Pro425 Asn122 | His435 Asp437 Arg423 Met424 Asn118 Ser422 | Asp437 Arg423 Ser422 Pro425 Asn118 | Arg423 Try75 Gln71 Pro425 Met424 | Try75 Arg423 Ile426 | Arg423 Tyr75 Pro425 Tyr75 | Asn118 Tyr75 Asn118 |
| Hydrophobic interactions | Met424 Leu69 Phe119 | Arg423 Met424 Ala117 Phe119 | Ile426 Phe119Ala117 Leu69 Leu124 Ala117 | Ile426 Pro425Met424 Arg423 Leu69 Tyr75 | Leu69 Tyr75 Pro425Met424 Ile426 Pro425 Ile426 Ala117 | Leu69 Tyr75 Ile426 | Phe119 Ala117 Leu69Met111 Tyr75 |
| Van der Waals forces | Ile426 Lys445 Tyr123 Leu124 Met424 Pro425 Tyr75 Leu69 Met111 Gly112 | Tyr427 Leu323 Gln440 Pro425 Leu120 Leu69 Gln71 Asn122 Ala436 | Asp434 Lys445 Asn118 Met424 Ile426 Tyr75 Thr68 Tyr427 Tyr123 Leu69 Gln440 Asn112 Phe119 Arg63 | Gln440 Ser422 Asp434 His435 Ala436 Thr68 Leu323 Tyr427 | Gln71 Thr68 Met111 Ala67 Arg63 Ser422 Asp437 Tyr427 Asn118 Phe119 Thr68 | Tyr453 Pro425 Gly452 Met424 Thr68 Gln71 Ala117 Tyr427 | Leu274 Leu120 Ala113 Gly116 Gly112 Asp72 Thr74 Ala67 Tyr75 Arg63 Gln71 Thr70 |
| Electrostatic interactions | — | — | Asp72 | Met424Asp437 | Arg423 | — | — |
| (kcal/mol) | RK7 | KQ7 | QP13 | VN10 |
|---|---|---|---|---|
| VDWAALS | −89.74 ± 0.01 | −74.74 ± 1.38 | −84.13 ± 2.79 | −68.20 ± 0.15 |
| ΔEEL | −25.73 ± 3.86 | −72.67 ± 3.27 | −50.58 ± 7.24 | −152.73 ± 1.87 |
| ΔEGB | 65.11 ± 1.11 | 91.60 ± 3.06 | 106.08 ± 7.32 | 229.91 ± 7.28 |
| ΔEsurf | −10.99 ± 0.01 | −9.44 ± 0.20 | −11.34 ± 0.02 | −10.06 ± 0.11 |
| ΔGgas | −115.46 ± 3.86 | −147.40 ± 3.55 | −134.71 ± 7.75 | −220.94 ± 1.87 |
| ΔGsolv | 54.12 ± 1.11 | 82.16 ± 3.07 | 94.74 ± 7.32 | 219.84 ± 7.28 |
| ΔTotal | −61.34 ± 4.02 | −65.24 ± 4.69 | −39.97 ± 10.66 | −1.09 ± 7.52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Song, X.; Liang, Q. Study on the Inhibitory Effect of Bioactive Peptides Derived from Yak Milk Cheese on Cholesterol Esterase. Foods 2024, 13, 2970. https://doi.org/10.3390/foods13182970
Wang P, Song X, Liang Q. Study on the Inhibitory Effect of Bioactive Peptides Derived from Yak Milk Cheese on Cholesterol Esterase. Foods. 2024; 13(18):2970. https://doi.org/10.3390/foods13182970
Chicago/Turabian StyleWang, Peng, Xuemei Song, and Qi Liang. 2024. "Study on the Inhibitory Effect of Bioactive Peptides Derived from Yak Milk Cheese on Cholesterol Esterase" Foods 13, no. 18: 2970. https://doi.org/10.3390/foods13182970