
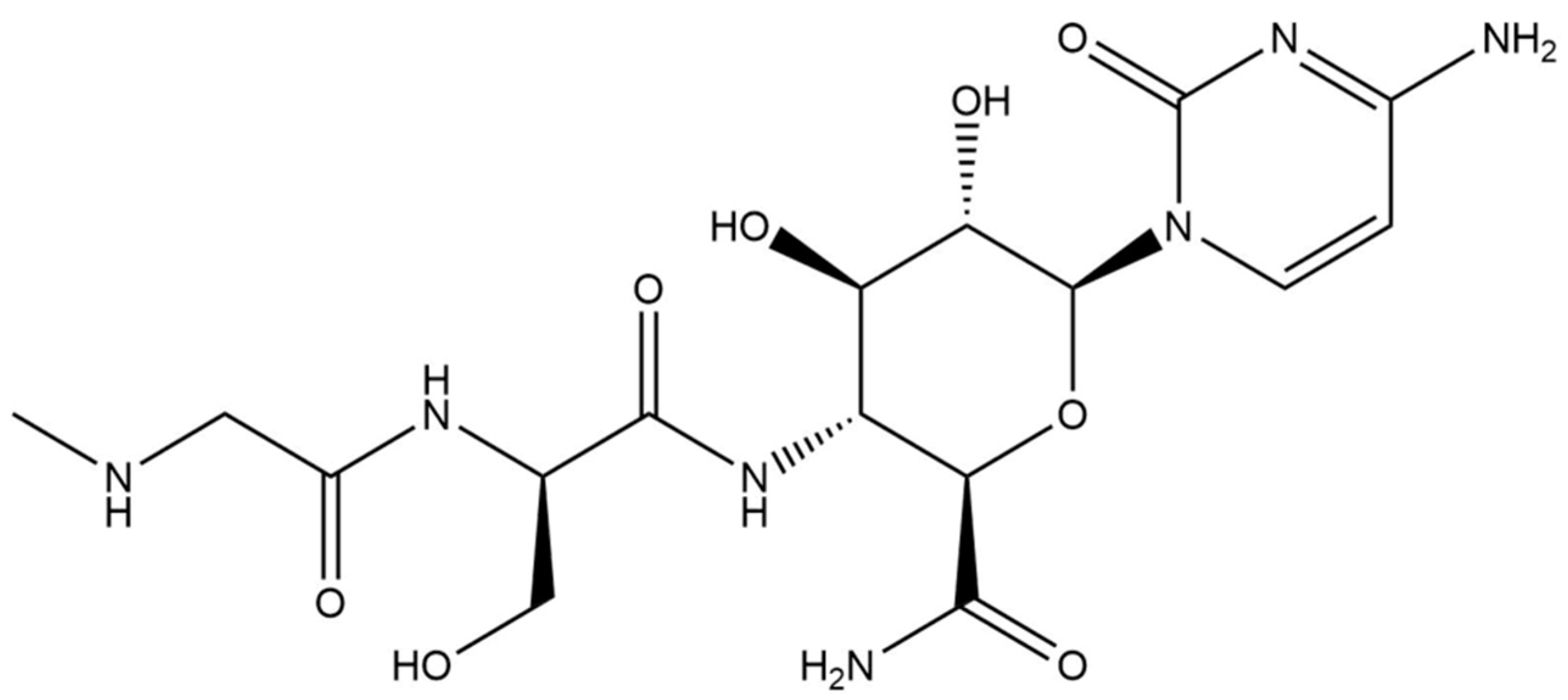
Development of a Cation Exchange SPE-HILIC-MS/MS Method for the Determination of Ningnanmycin Residues in Tea and Chrysanthemum


Abstract
1. Introduction
2. Materials and Methods
2.1. Analytical Instruments
2.2. Reagents and Materials
2.3. Preparation of the Standard Solution
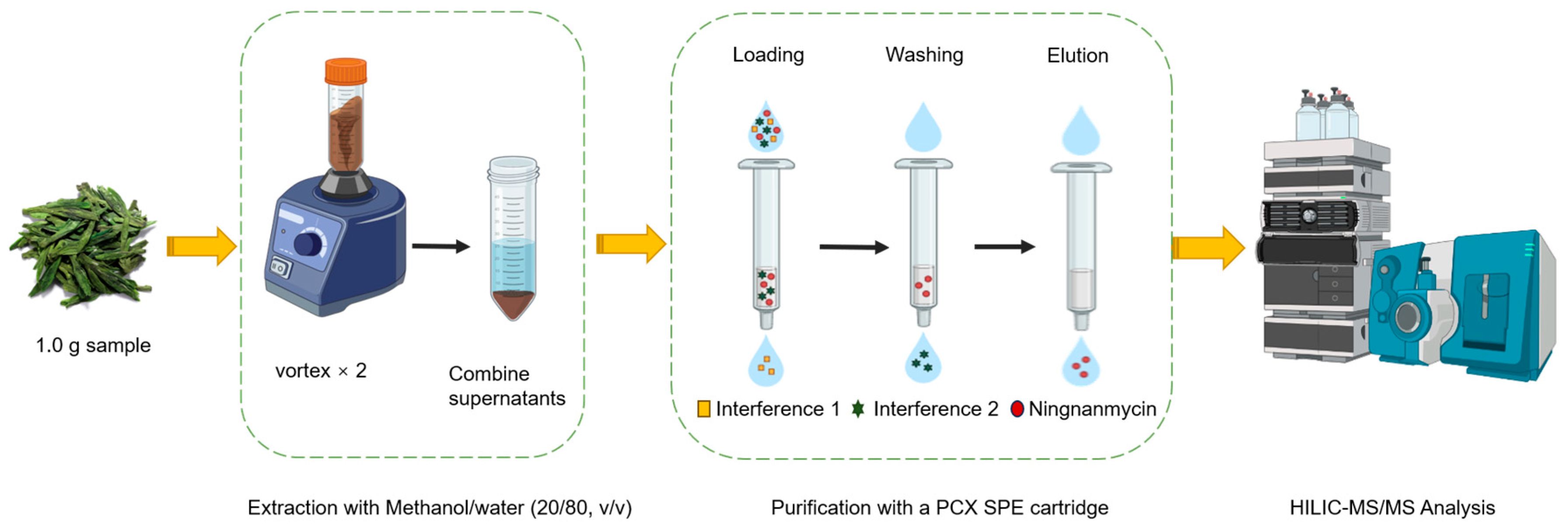
2.4. Sample Pretreatment
2.5. Instrumental Conditions
2.6. Matrix Effect
3. Results and Discussion
3.1. Optimization of Instrument Parameters
3.1.1. Optimization of MS/MS Parameters
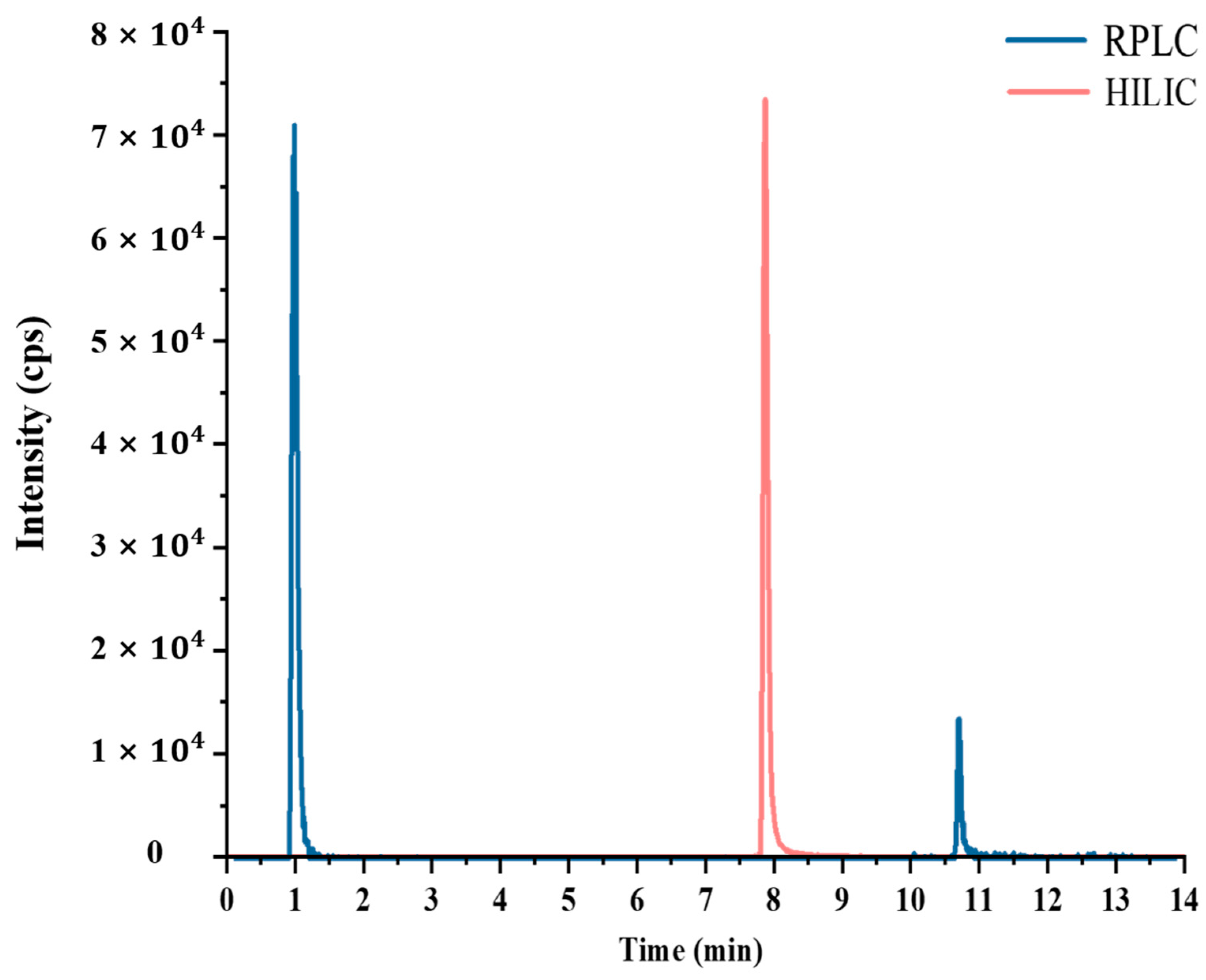
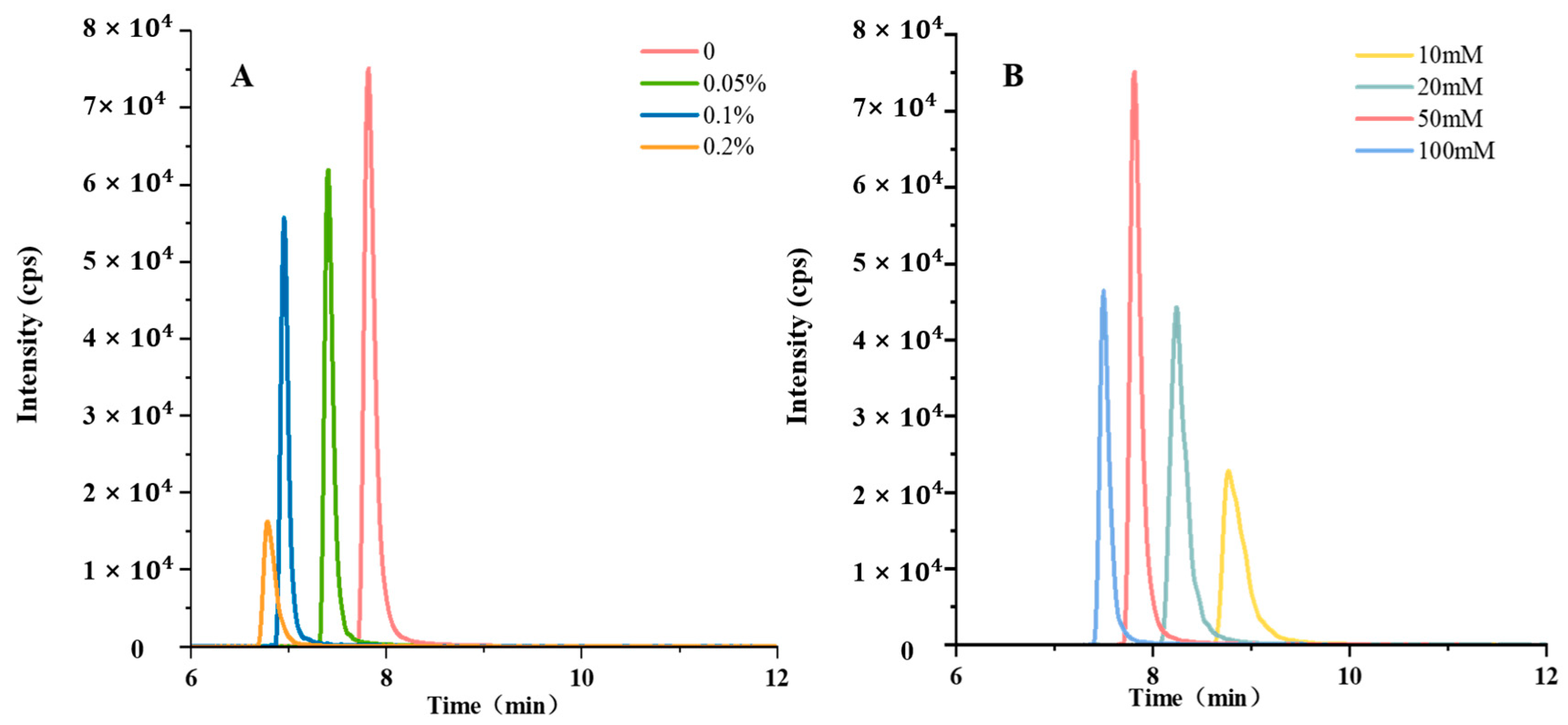
3.1.2. Optimization of Chromatographic Conditions
3.2. Optimization of Sample Preparation
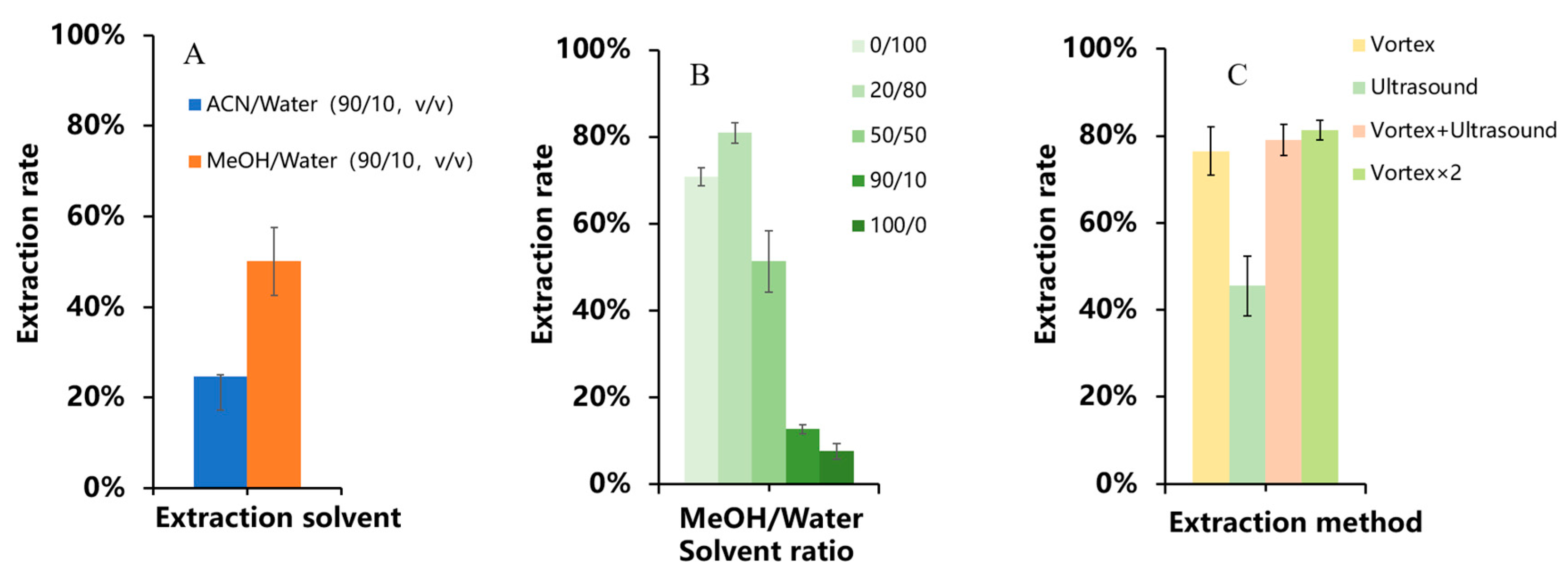
3.2.1. Optimization of Extraction Conditions
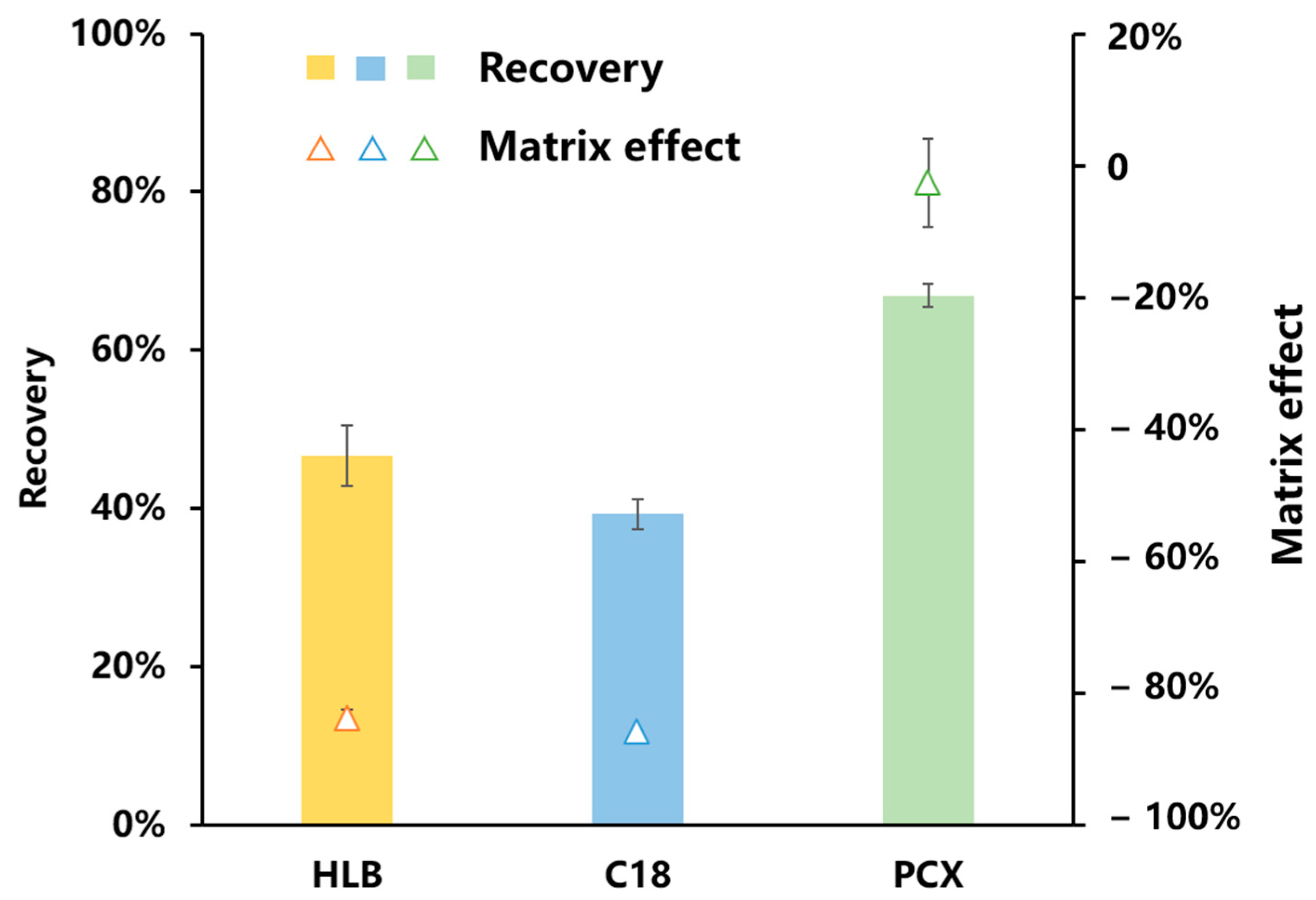
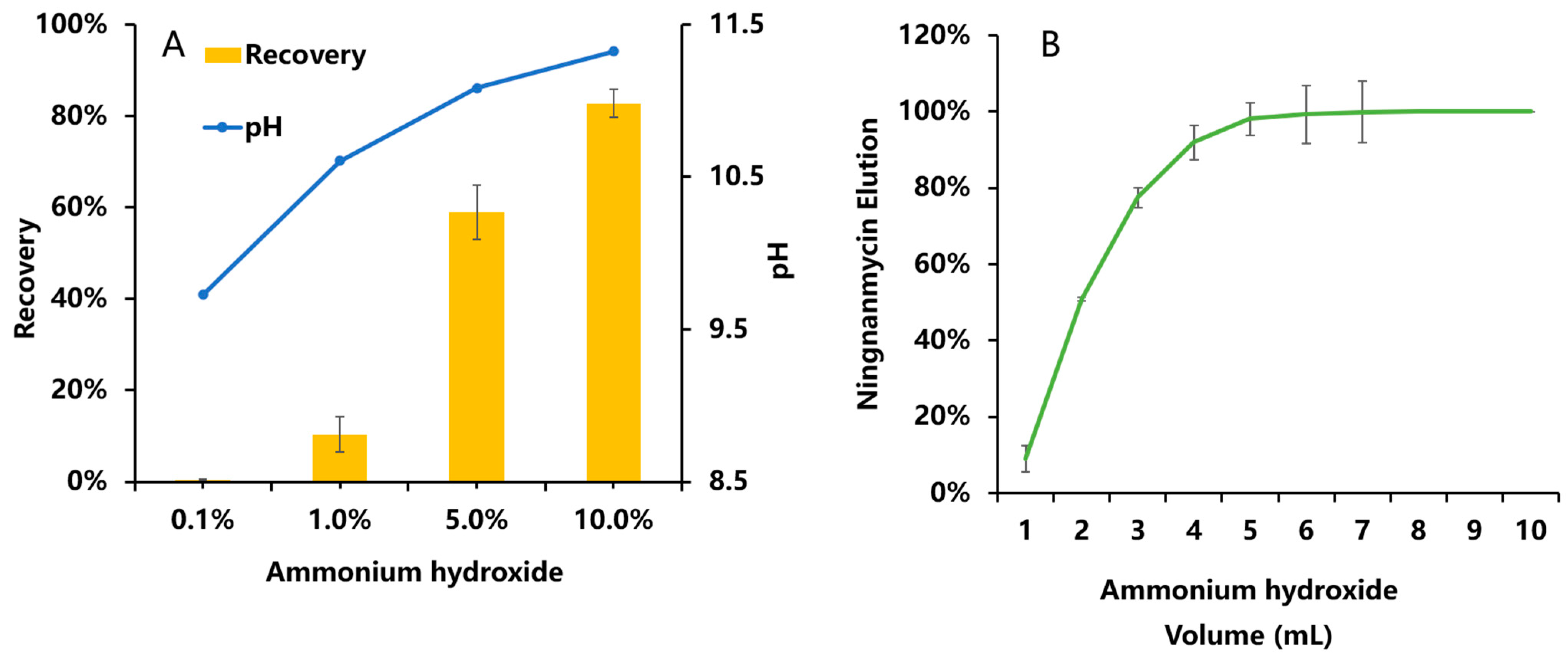
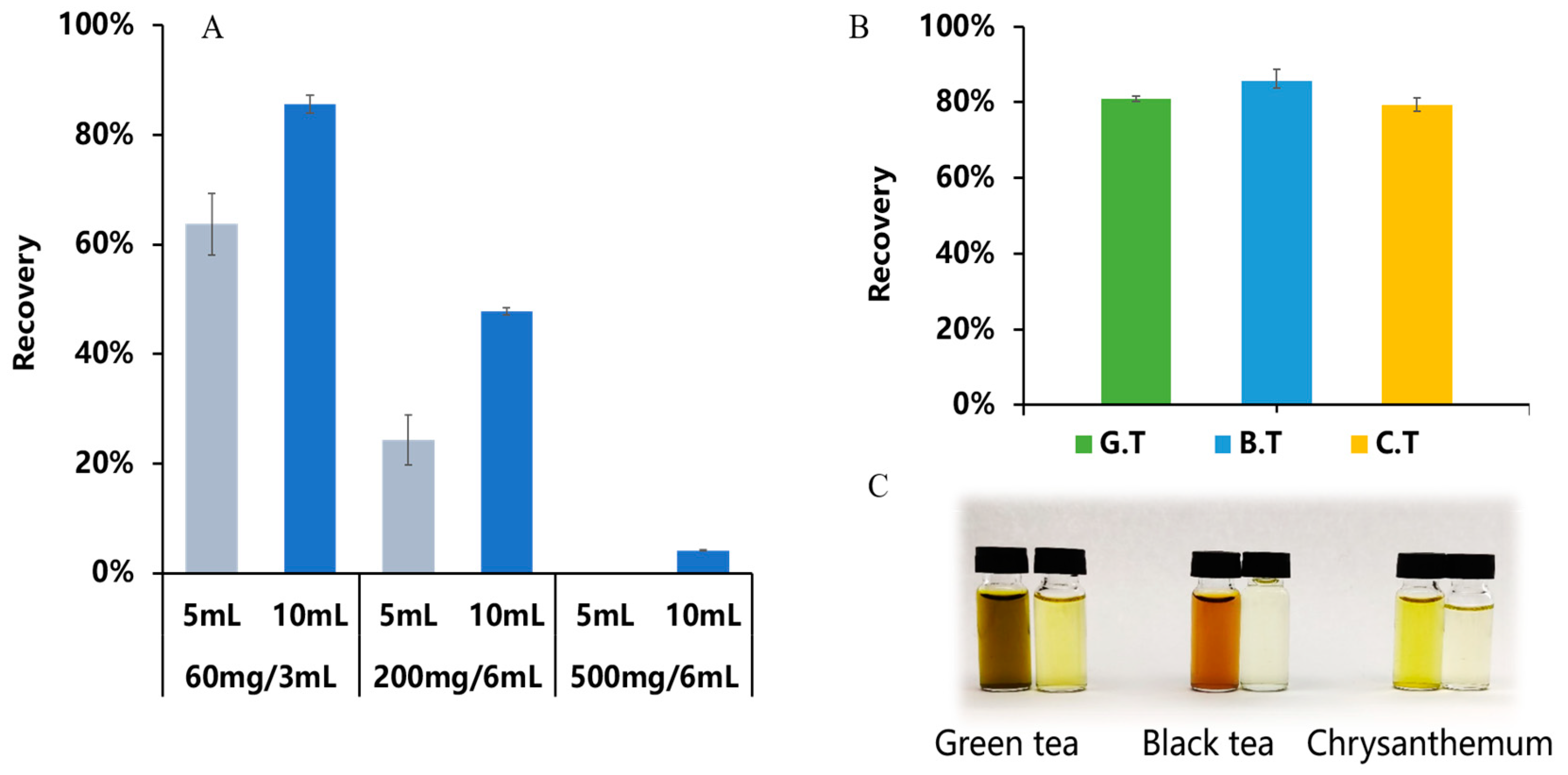
3.2.2. Optimization of the Purification Conditions
3.3. Method Validation
3.3.1. Accuracy and Precision
3.3.2. Sensitivity
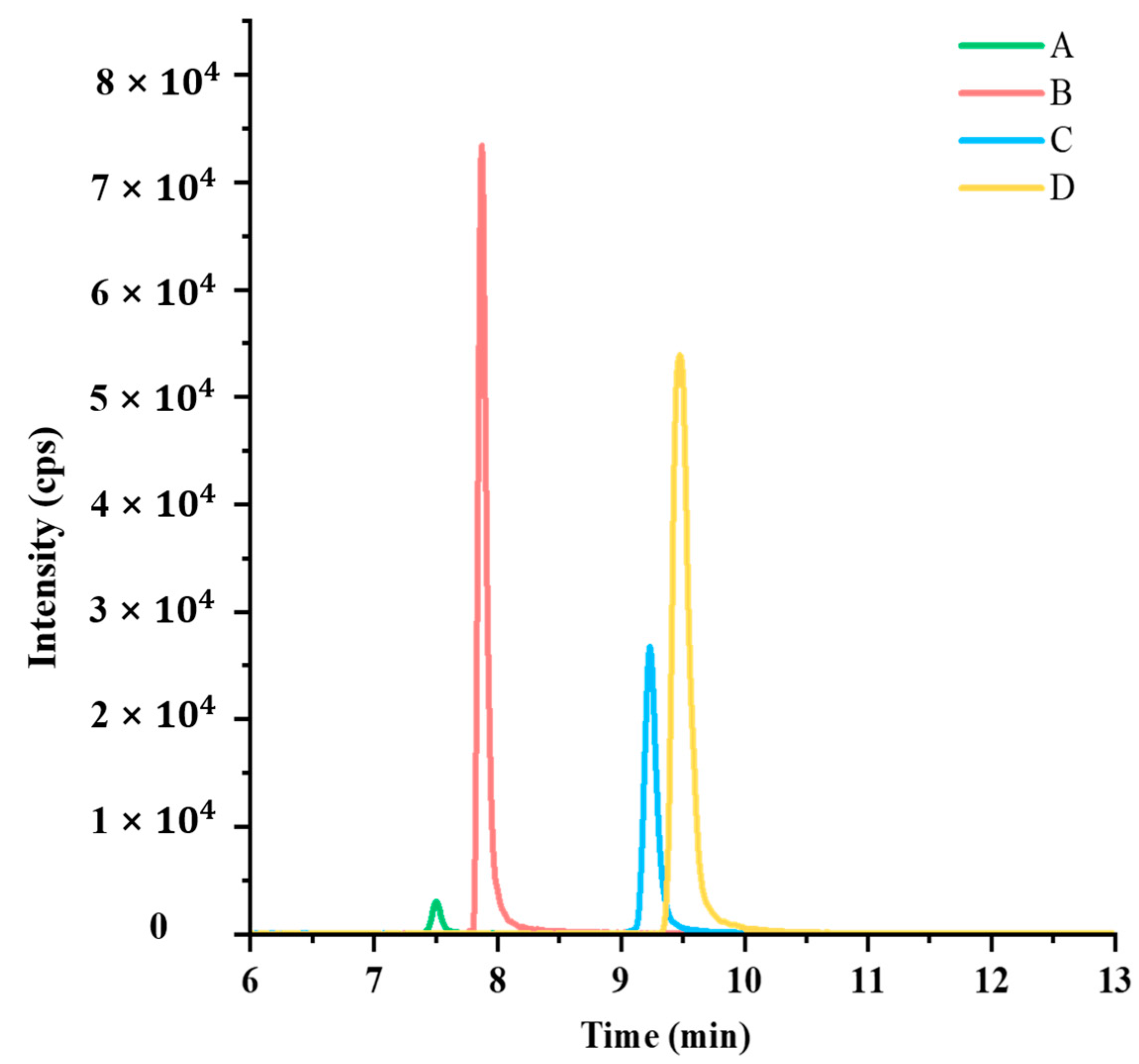
3.3.3. Linearity and Matrix Effect
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Han, Y.; Luo, Y.; Qin, S.; Xi, L.; Wan, B.; Du, L. Induction of systemic resistance against tobacco mosaic virus by Ningnanmycin in tobacco. Pest. Biochem. Physiol. 2014, 111, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Jiang, S.; Wen, X.; Song, X.; Wang, X.; Li, D.; Yin, Q.; Wu, X.; Wang, D.; Chen, Z. Antifungal activity and le mode of action of ningnanmycin against tea gray blight disease pathogen Pseudopestalotiopsis camelliae-sinensis. Pathology 2021, 111, 1735–1742. [Google Scholar] [CrossRef]
- GB 2763-2021; National Food Safety Standard-Maximum Residue Limits For Pesticides In Food. National Health Commission of the P.R.C., Ministry of Agriculture and Rural Affairs of the P.R.C., State Administration for Market Regulation: Beijing, China, 2021; Volume 7, p. 254.
- Iturrospe, E.; Da Silva, K.M.; Andújar, B.T.; Cuykx, M.; Boeckmans, J.; Vanhaecke, T.; Covaci, A.; Van Nuijs, A.L. An atory approach for an oriented development of an untargeted hydrophilic interaction liquid chromatography-mass spectry platform for polar metabolites in biological matrices. J. Chromatogr. A 2021, 1637, 461807. [Google Scholar] [CrossRef] [PubMed]
- Periat, A.; Kohler, I.; Bugey, A.; Bieri, S.; Versace, F.; Staub, C.; Guillarme, D. Hydrophilic interaction chromatography versus reversed phase liquid chromatography coupled to mass spectrometry: Effect of electrospray ionization source geometry on sensitivity. J. Chromatogr. A 2014, 1356, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Buszewski, B.; Noga, S. Hydrophilic interaction liquid chromatography (HILIC)—A powerful separation technique. Bioanal. Chem. 2012, 402, 231–247. [Google Scholar] [CrossRef]
- Wang, T.; Yin, S.; Hu, J. Dissipation and residue determination of ningnanmycin in cucumber and soil by high perfor liquid chromatography with ultraviolet detector. Bull. Environ. Contam. Toxicol. 2013, 90, 256–259. [Google Scholar] [CrossRef]
- Li, W.; Dai, X.; Pu, E.; Bian, H.; Chen, Z.; Zhang, X.; Guo, Z.; Li, P.; Li, H.; Yong, Y.; et al. HLB-MCX-Based Solid-Phase tion Combined with Liquid Chromatography-Tandem Mass Spectrometry for the Simultaneous Determination of Four ultural Antibiotics (Kasugamycin, Validamycin A, Ningnanmycin, and Polyoxin B) Residues in Plant-Origin Foods. J. Agric. Food Chem. 2020, 68, 14025–14037. [Google Scholar] [CrossRef]
- Zongmao, C.; Haibin, W. Factors affecting residues of pesticides in tea. Pestic. Sci. 1988, 23, 109–118. [Google Scholar] [CrossRef]
- Narenderan, S.T.; Meyyanathan, S.N.; Babu, B. Review of pesticide residue analysis in fruits and vegetables. Pre-treatextraction and detection techniques. Food Res. Int. 2020, 133, 109141. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, S.; Cui, X.; Pan, C.; Zhang, A.; Chen, F. A review of sample preparation methods for the pesticide residue is in foods. Cent. Eur. J. Chem. 2012, 10, 900–925. [Google Scholar] [CrossRef]
- Song, X.; Li, F.; Yan, T.; Tian, F.; Ren, L.; Jiang, C.; Wang, Q.; Zhang, S. Research progress in the sample pretreatment ques and advanced quick detection methods of pesticide residues. Process Saf. Environ. Protect. 2022, 165, 610–622. [Google Scholar] [CrossRef]
- Artymowicz, M.; Macioszek, S.; Jacyna, J.; Dawidowska, J.; Struck-Lewicka, W.; Markuszewski, M.J.; Siluk, D. Developand optimization of modified nucleosides and deoxynucleosides simultaneous extraction with the use of Design of Exents approach. Sustain. Chem. Pharm. 2021, 21, 100439. [Google Scholar] [CrossRef]
- Socas-Rodriguez, B.; Herrera-Herrera, A.V.; Asensio-Ramos, M.; Hernandez-Borges, J. Recent applications of carbon ube sorbents in analytical chemistry. J. Chromatogr. A 2014, 1357, 110–146. [Google Scholar] [CrossRef]
- Kruve, A.; Künnapas, A.; Herodes, K.; Leito, I. Matrix effects in pesticide multi-residue analysis by liquid chromatog–mass spectrometry. J. Chromatogr. A 2008, 1187, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Yang, S.; Wang, P.G. Matrix effects and application of matrix effect factor. Future Sci. 2017, 9, 844. [Google Scholar] [CrossRef]
- Kruve, A.; Rebane, R.; Kipper, K.; Oldekop, M.; Evard, H.; Herodes, K.; Ravio, P.; Leito, I. Tutorial review on validation quid chromatography–mass spectrometry methods: Part I. Anal. Chim. Acta. 2015, 870, 29–44. [Google Scholar] [CrossRef]
- Mol, H.G.; Zomer, P.; López, M.G.; Fussell, R.J.; Scholten, J.; de Kok, A.; Wolheim, A.; Anastassiades, M.; Lozano, A.F. Identification in residue analysis based on liquid chromatography with tandem mass spectrometry: Experimental nce to update performance criteria. Anal. Chim. Acta. 2015, 873, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.Q.; Zou, L.; Yin, X.X.; Ong, C.N. HILIC-MS for metabolomics: An attractive and complementary approach to -MS. Mass Spectrom. Rev. 2016, 35, 574–600. [Google Scholar] [CrossRef]
- Žuvela, P.; Skoczylas, M.; Jay Liu, J.; Ba̧czek, T.; Kaliszan, R.; Wong, M.W.; Buszewski, B. Column characterization and on systems in reversed-phase high-performance liquid chromatography. Chem. Rev. 2019, 119, 3674–3729. [Google Scholar] [CrossRef] [PubMed]
- Jandera, P.; Hájek, T. Mobile phase effects on the retention on polar columns with special attention to the dual hydro interaction–reversed-phase liquid chromatography mechanism, a review. J. Sep. Sci. 2018, 41, 145–162. [Google Scholar] [CrossRef]
- Jandera, P.; Janás, P. Recent advances in stationary phases and understanding of retention in hydrophilic interaction atography. A review. Anal. Chim. Acta. 2017, 967, 12–32. [Google Scholar] [CrossRef] [PubMed]
- Gosetti, F.; Mazzucco, E.; Zampieri, D.; Gennaro, M.C. Signal suppression/enhancement in high-performance liquid atography tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 3929–3937. [Google Scholar] [CrossRef] [PubMed]
- Jandera, P. Stationary and mobile phases in hydrophilic interaction chromatography: A review. Anal. Chim. Acta. 2011, 692, 1–25. [Google Scholar] [CrossRef] [PubMed]
- McCalley, D.V. Is hydrophilic interaction chromatography with silica columns a viable alternative to reversed-phase chromatography for the analysis of ionisable compounds? J. Chromatogr. A 2007, 1171, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Yang, W.; Li, Y.; Sun, C. Multi-residue analytical methods for pesticides in teas: A review. Eur. Food Res. Technol. 2021, 247, 1839–1858. [Google Scholar] [CrossRef]
- Anastassiades, M.; Kolberg, D.I.; Eichhorn, E.; Wachtler, A.K.; Benkenstein, A.; Zechmann, S.; Mack, D.; Wildgrube, C.A.; Sigalov, I. Quick Method for the Analysis of Numerous Highly Polar Pesticides in Food Involving Extraction with Acidified Methanol and LC-MS/MS Measurement I. Food of Plant Origin (QuPPe-PO-Method), EU Reference Laboratory for Pesticides Requiring Single Residue Methods (EURL-SRM) Version 12.1, 20230317, 99. Available online: https://www.quppe.eu/quppe_doc.asp (accessed on 31 December 2023).
- Hansen, S.H.; Pedersen-Bjergaard, S. Bioanalysis of Pharmaceuticals: Sample Preparation, Separation Techniques and Spectrometry; Wiley: Hoboken, NJ, USA, 2015; pp. 73–122. ISBN 9781118716861. [Google Scholar]
- Pérez-Rodríguez, M.; Pellerano, R.G.; Pezza, L.; Pezza, H.R. An overview of the main foodstuff sample preparation ologies for tetracycline residue determination. Talanta 2018, 182, 1–21. [Google Scholar] [CrossRef]
- Nasiri, A.; Jahani, R.; Mokhtari, S.; Yazdanpanah, H.; Daraei, B.; Faizi, M.; Kobarfard, F. Overview, consequences, and gies for overcoming matrix effects in LC-MS analysis: A critical review. Analyst 2021, 146, 6049–6063. [Google Scholar] [CrossRef] [PubMed]
- Hajšlová, J.; Zrostlıkova, J. Matrix effects in (ultra) trace analysis of pesticide residues in food and biotic matrices. J. Matogr. A 2003, 1000, 181–197. [Google Scholar] [CrossRef]
- Kaczyński, P. Clean-up and matrix effect in LC-MS/MS analysis of food of plant origin for high polar herbicides. Food 2017, 230, 524–531. [Google Scholar] [CrossRef]
- Uhrovčík, J. Strategy for determination of LOD and LOQ values–Some basic aspects. Talanta 2014, 119, 178–180. [Google Scholar] [CrossRef]
- Tomšič, R.; Heath, D.; Heath, E.; Markelj, J.; Kandolf Borovšak, A.; Prosen, H. Determination of neonicotinoid pesticides in propolis with liquid chromatography coupled to tandem mass spectrometry. Molecules 2020, 25, 5870. [Google Scholar] [CrossRef] [PubMed]
- Kittlaus, S.; Schimanke, J.; Kempe, G.; Speer, K. Assessment of sample cleanup and matrix effects in the pesticide residue analysis of foods using postcolumn infusion in liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2011, 1218, 8399–8410. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tea Samples | Recoveries (%) | Interday Precision (RSD, %) | Intraday Precision (RSD, %) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Spiked Level (μg/kg) | Spiked Level (μg/kg) | Spiked Level (μg/kg) | |||||||
| C1 | C2 | C3 | C1 | C2 | C3 | C1 | C2 | C3 | |
| G.T | 77.3 | 78.4 | 82.0 | 4.9 | 6.9 | 3.2 | 4.2 | 5.1 | 0.5 |
| B.T | 80.1 | 80.5 | 81.5 | 3.9 | 7.0 | 3.6 | 1.0 | 0.6 | 0.7 |
| C.T | 74.0 | 80.0 | 75.3 | 5.8 | 7.7 | 4.1 | 2.5 | 1.5 | 0.6 |
| Tea Samples | Calibration Equation | R2 | Linear Range | LOQ | LOD | ME |
|---|---|---|---|---|---|---|
| (μg/L) | (μg/kg) | (μg/kg) | (%) | |||
| G.T | y = 706.06x + 1055.7 | 0.9998 | 1–1000 | 3.6 | 1.1 | 18.9 |
| B.T | y = 621.24x + 1496.5 | 0.9995 | 1–1000 | 11.3 | 3.4 | 1.6 |
| C.T | y = 735.08x + 1418.1 | 0.9985 | 1–1000 | 23.7 | 7.1 | 23.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, A.; Wang, C.; Wu, Z.; Liu, Y.; Hao, Z.; Lu, C.; Chen, H. Development of a Cation Exchange SPE-HILIC-MS/MS Method for the Determination of Ningnanmycin Residues in Tea and Chrysanthemum. Foods 2024, 13, 635. https://doi.org/10.3390/foods13050635
Li A, Wang C, Wu Z, Liu Y, Hao Z, Lu C, Chen H. Development of a Cation Exchange SPE-HILIC-MS/MS Method for the Determination of Ningnanmycin Residues in Tea and Chrysanthemum. Foods. 2024; 13(5):635. https://doi.org/10.3390/foods13050635
Chicago/Turabian StyleLi, Aiping, Chen Wang, Zhenghao Wu, Yingying Liu, Zhenxia Hao, Chengyin Lu, and Hongping Chen. 2024. "Development of a Cation Exchange SPE-HILIC-MS/MS Method for the Determination of Ningnanmycin Residues in Tea and Chrysanthemum" Foods 13, no. 5: 635. https://doi.org/10.3390/foods13050635
APA StyleLi, A., Wang, C., Wu, Z., Liu, Y., Hao, Z., Lu, C., & Chen, H. (2024). Development of a Cation Exchange SPE-HILIC-MS/MS Method for the Determination of Ningnanmycin Residues in Tea and Chrysanthemum. Foods, 13(5), 635. https://doi.org/10.3390/foods13050635