
A Fast and Simple Solid Phase Extraction-Based Method for Glucosinolate Determination: An Alternative to the ISO-9167 Method
 ,
,
Abstract
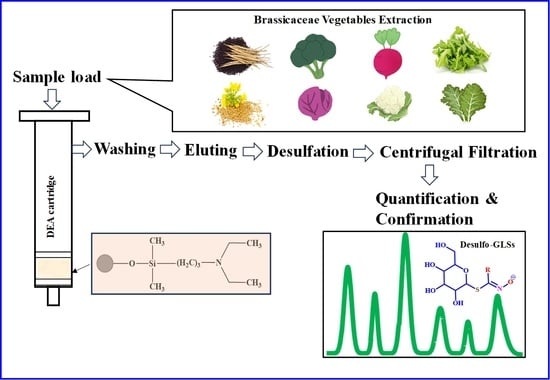
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Sample Information
2.3. Glucosinolates Extraction
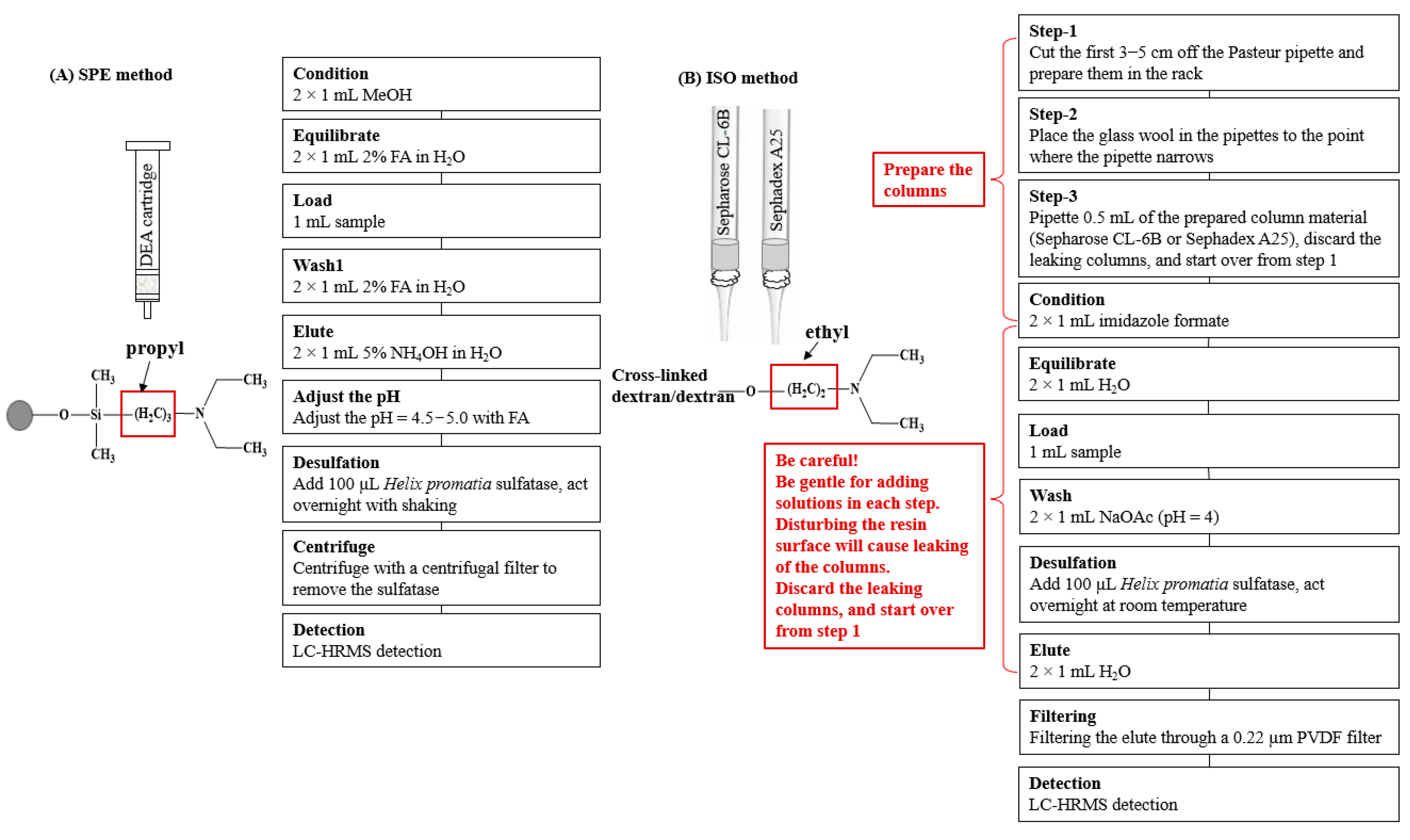
2.4. SPE Cartridges
2.5. Purification of Glucosinolates
2.6. Desulfation of Glucosinolates
2.7. Purification and Desulfation of Glucosinolates with the ISO Method
2.8. UHPLC-HRMS Conditions for the Detection of Glucosinolates
2.9. Quantification of the Total and Individual Glucosinolates
2.10. Statistical Analysis
3. Results and Discussion
3.1. Selection of Different Commercial Ion-Exchange SPE Cartridges
3.2. Method Optimization
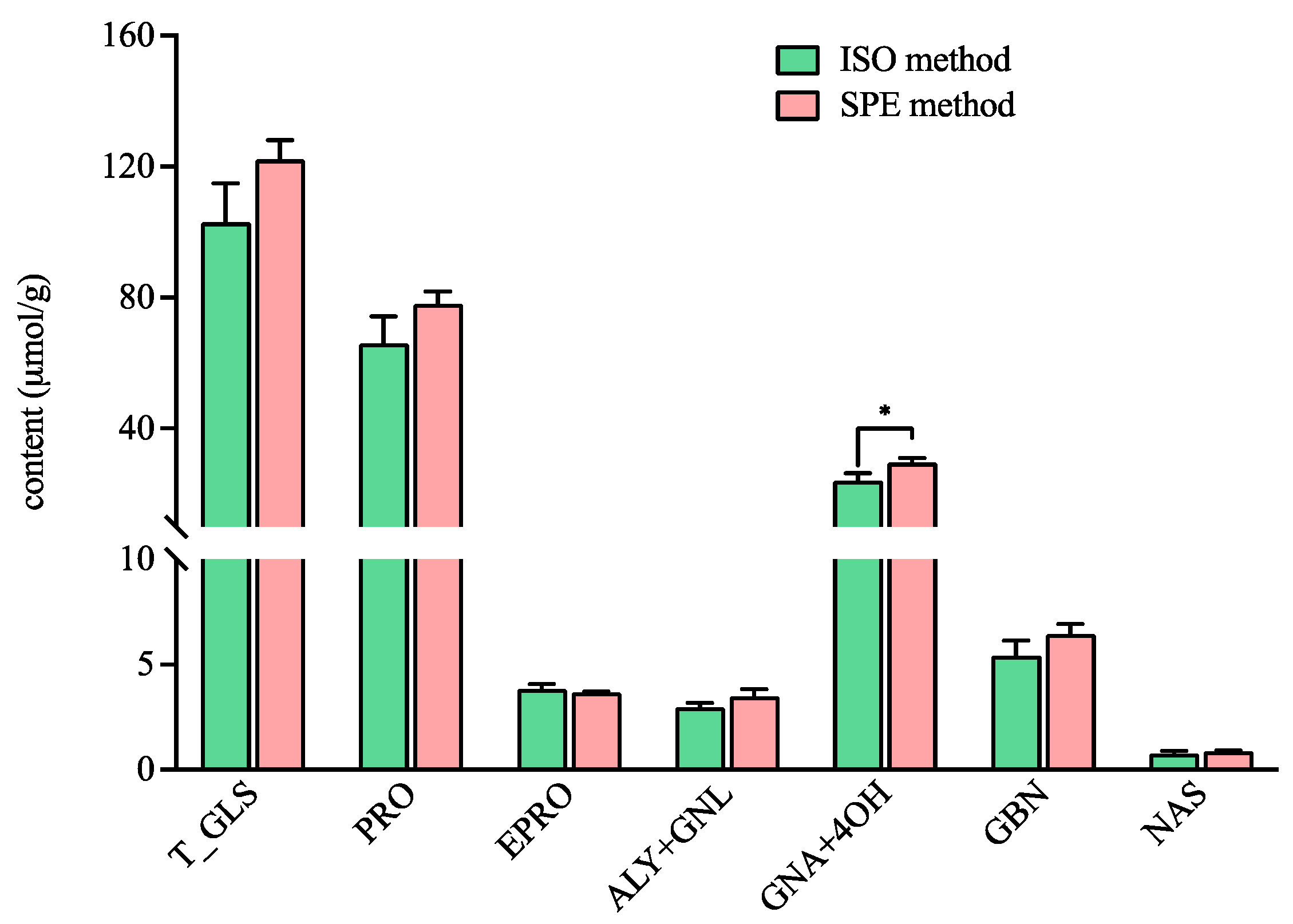
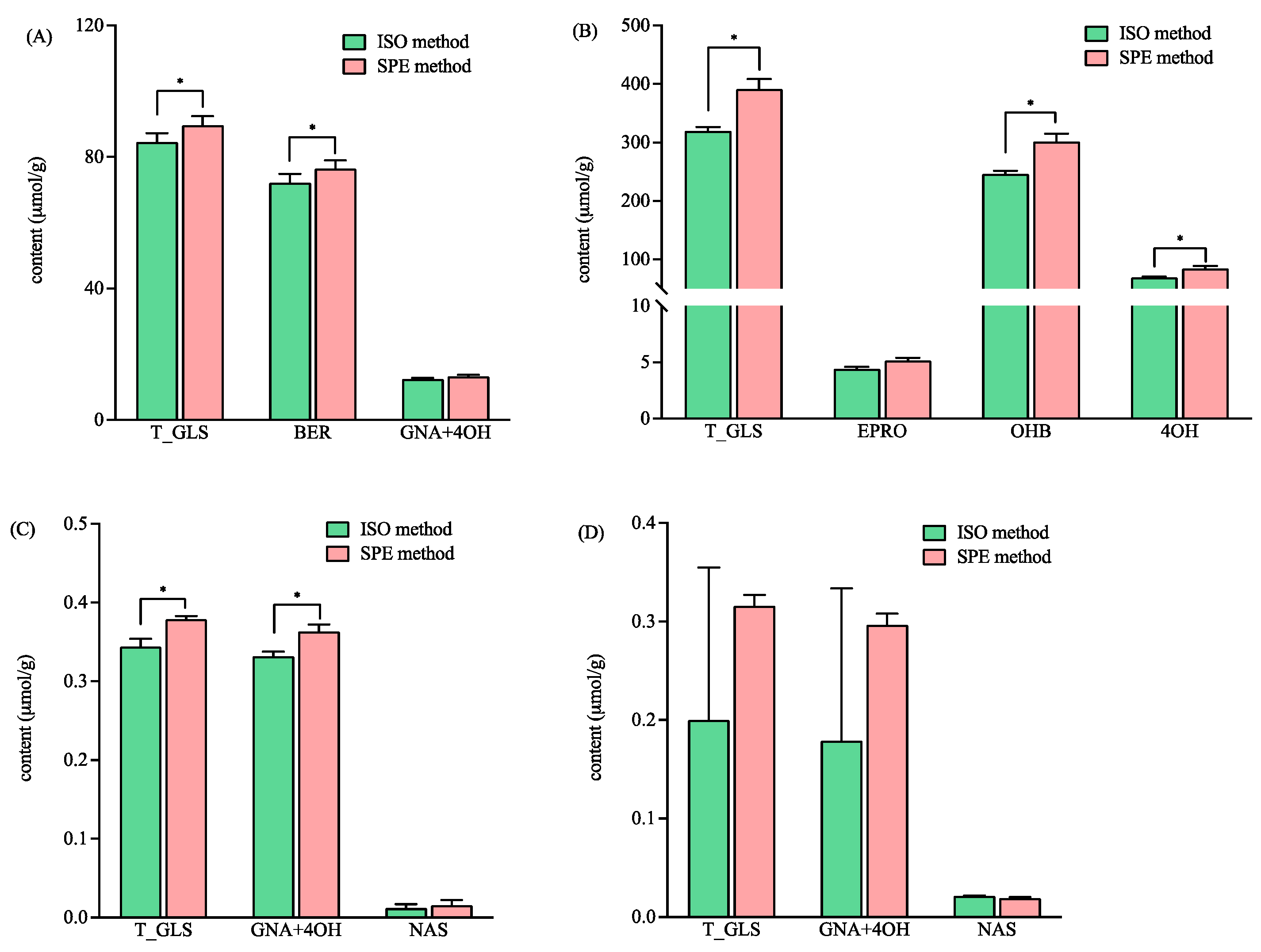
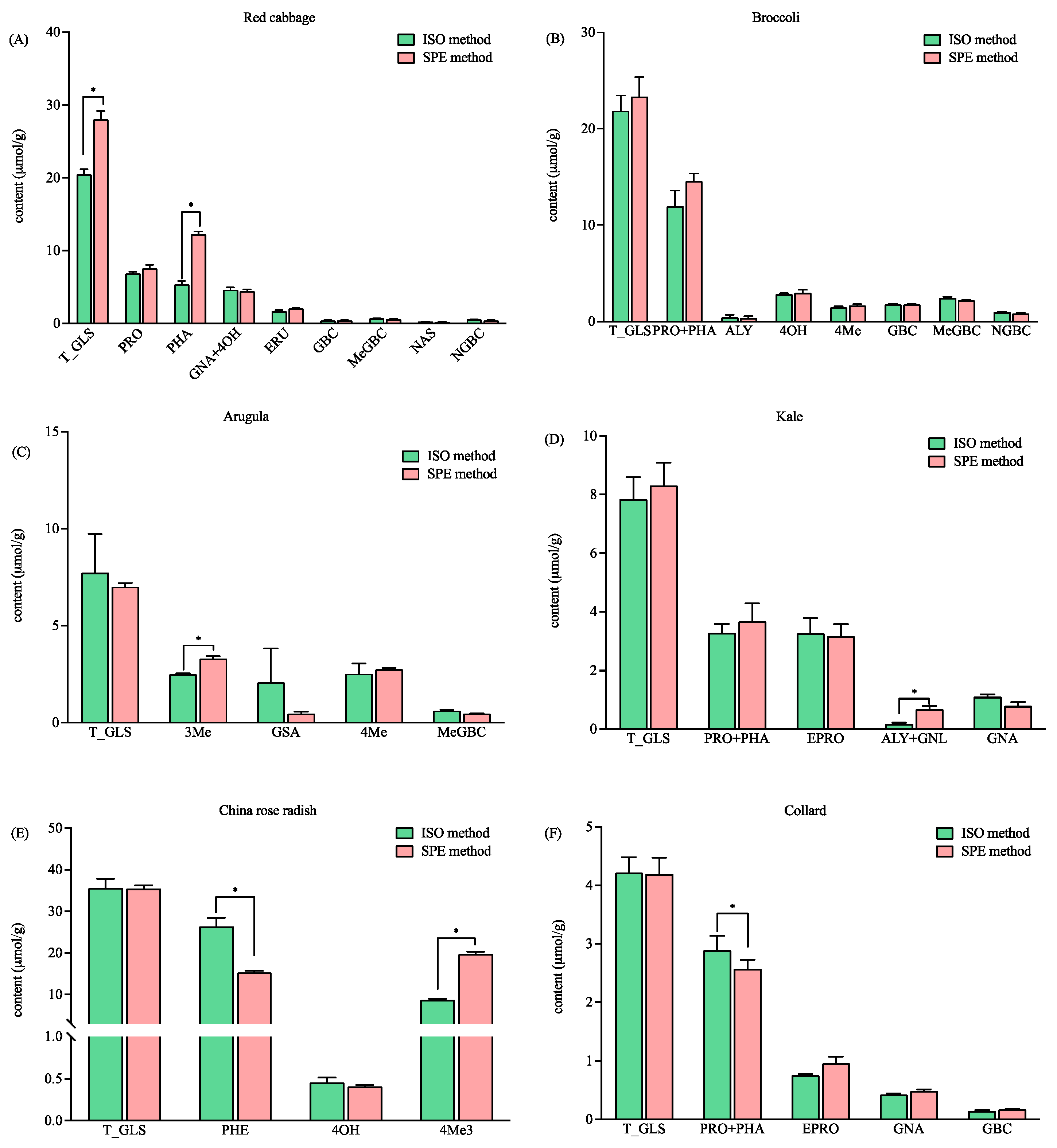
3.3. Application in Brassicaceae Vegetables Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sikorska-Zimny, K.; Beneduce, L. The glucosinolates and their bioactive derivatives in Brassica: A review on classification, biosynthesis and content in plant tissues, fate during and after processing, effect on the human organism and interaction with the gut microbiota. Crit. Rev. Food Sci. Nutr. 2021, 61, 2544–2571. [Google Scholar] [CrossRef]
- Halkier, B.A.; Gershenzon, J. Biology and biochemistry of glucosinolates. Annu. Rev. Plant Biol. 2006, 57, 303–333. [Google Scholar] [CrossRef]
- Clarke, D.B. Glucosinolates, structures and analysis in food. Anal. Methods 2010, 2, 310–325. [Google Scholar] [CrossRef]
- Blažević, I.; Montaut, S.; Burčul, F.; Olsen, C.E.; Burow, M.; Rollin, P.; Agerbirk, N. Glucosinolate structural diversity, identification, chemical synthesis and metabolism in plants. Phytochemistry 2020, 169, 112100–112156. Available online: https://www.sciencedirect.com/science/article/pii/S003194221930069X (accessed on 18 January 2024). [CrossRef]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef]
- Bosetti, C.; Filomeno, M.; Riso, P.; Polesel, J.; Levi, F.; Talamini, R.; Montella, M.; Negri, E.; Franceschi, S.; La Vecchia, C. Cruciferous vegetables and cancer risk in a network of case–control studies. Ann. Oncol. 2012, 23, 2198–2203. [Google Scholar] [CrossRef]
- Long, J.; Liu, Z.; Liang, S.; Chen, B. Cruciferous Vegetable Intake and Risk of Prostate Cancer: A Systematic Review and Meta-Analysis. Urol. Int. 2023, 107, 723–733. [Google Scholar] [CrossRef]
- Azeem, S.; Gillani, S.W.; Siddiqui, A.; Jandrajupalli, S.B.; Poh, V.; Syed Sulaiman, S.A. Diet and Colorectal Cancer Risk in Asia--a Systematic Review. Asian Pac. J. Cancer Prev. APJCP 2015, 16, 5389–5396. [Google Scholar] [CrossRef]
- Hansson, L.E.; Nyrén, O.; Bergström, R.; Wolk, A.; Lindgren, A.; Baron, J.; Adami, H.O. Diet and risk of gastric cancer. A population-based case-control study in Sweden. Int. J. Cancer 1993, 55, 181–189. [Google Scholar] [CrossRef]
- Al-Zalabani, A.H.; Stewart, K.F.; Wesselius, A.; Schols, A.M.; Zeegers, M.P. Modifiable risk factors for the prevention of bladder cancer: A systematic review of meta-analyses. Eur. J. Epidemiol. 2016, 31, 811–851. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, L. Cruciferous vegetables intake is associated with lower risk of renal cell carcinoma: Evidence from a meta-analysis of observational studies. PLoS ONE 2013, 8, e75732. [Google Scholar] [CrossRef]
- Wu, Q.J.; Yang, G.; Zheng, W.; Li, H.L.; Gao, J.; Wang, J.; Gao, Y.T.; Shu, X.O.; Xiang, Y.B. Pre-diagnostic cruciferous vegetables intake and lung cancer survival among Chinese women. Sci. Rep. 2015, 5, 10306. [Google Scholar] [CrossRef]
- Kamal, R.M.; Abdull Razis, A.F.; Mohd Sukri, N.S.; Perimal, E.K.; Ahmad, H.; Patrick, R.; Djedaini-Pilard, F.; Mazzon, E.; Rigaud, S. Beneficial Health Effects of Glucosinolates-Derived Isothiocyanates on Cardiovascular and Neurodegenerative Diseases. Molecules 2022, 27, 624. [Google Scholar] [CrossRef]
- Rochfort, S.; Caridi, D.; Stinton, M.; Trenerry, V.C.; Jones, R. The isolation and purification of glucoraphanin from broccoli seeds by solid phase extraction and preparative high performance liquid chromatography. J. Chromatogr. A 2006, 1120, 205–210. [Google Scholar] [CrossRef]
- Tian, Q.; Rosselot, R.A.; Schwartz, S.J. Quantitative determination of intact glucosinolates in broccoli, broccoli sprouts, Brussels sprouts, and cauliflower by high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry. Anal. Biochem. 2005, 343, 93–99. [Google Scholar] [CrossRef]
- Doheny-Adams, T.; Redeker, K.; Kittipol, V.; Bancroft, I.; Hartley, S.E. Development of an efficient glucosinolate extraction method. Plant Methods 2017, 13, 17. [Google Scholar] [CrossRef]
- Glauser, G.; Schweizer, F.; Turlings, T.C.; Reymond, P. Rapid profiling of intact glucosinolates in Arabidopsis leaves by UHPLC-QTOFMS using a charged surface hybrid column. Phytochem. Anal. PCA 2012, 23, 520–528. [Google Scholar] [CrossRef]
- Crocoll, C.; Halkier, B.A.; Burow, M. Analysis and Quantification of Glucosinolates. Curr. Protoc. Plant Biol. 2016, 1, 385–409. [Google Scholar] [CrossRef]
- Ali Redha, A.; Langston, F.; Nash, G.R.; Bows, J.R.; Torquati, L.; Gidley, M.J.; Cozzolino, D. Determination of glucosinolates in broccoli (Brassica oleracea var. italica) by combining mid-infrared (MIR) spectroscopy with chemometrics. Int. J. Food Sci. Technol. 2023, 58, 5679–5688. [Google Scholar] [CrossRef]
- Aguilar-Galvez, A.; García-Ríos, D.; Lindo, J.; Ramírez-Guzmán, D.; Chirinos, R.; Pedreschi, R.; Campos, D. Impact of cold storage followed by drying of mashua tuber (Tropaeolum tuberosum) on the glucosinolate content and their transformation products. Int. J. Food Sci. Technol. 2022, 57, 7797–7805. [Google Scholar] [CrossRef]
- Omirou, M.; Papastylianou, I.; Iori, R.; Papastephanou, C.; Papadopoulou, K.K.; Ehaliotis, C.; Karpouzas, D.G. Microwave-assisted extraction of glucosinolates from Eruca sativa seeds and soil: Comparison with existing methods. Phytochem. Anal. 2009, 20, 214–220. [Google Scholar] [CrossRef]
- Wu, X.; Huang, H.; Childs, H.; Wu, Y.; Yu, L.; Pehrsson, P.R. Glucosinolates in Brassica Vegetables: Characterization and Factors That Influence Distribution, Content, and Intake. Annu. Rev. Food Sci. Technol. 2021, 12, 485–511. [Google Scholar] [CrossRef]
- Trenerry, V.C.; Caridi, D.; Elkins, A.; Donkor, O.; Jones, R. The determination of glucoraphanin in broccoli seeds and florets by solid phase extraction and micellar electrokinetic capillary chromatography. Food Chem. 2006, 98, 179–187. [Google Scholar] [CrossRef]
- Corporation, W. Beginner’s Guide to SPE: Solid-Phase Extraction; Waters Corporation: Milford, MA, USA, 2014. [Google Scholar]
- Nakatani, K.; Izumi, Y.; Takahashi, M.; Bamba, T. Unified-Hydrophilic-Interaction/Anion-Exchange Liquid Chromatography Mass Spectrometry (Unified-HILIC/AEX/MS): A Single-Run Method for Comprehensive and Simultaneous Analysis of Polar Metabolome. Anal. Chem. 2022, 94, 16877–16886. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, M.; Chen, P. GLS-Finder: A Platform for Fast Profiling of Glucosinolates in Brassica Vegetables. J. Agric. Food Chem. 2016, 64, 4407–4415. [Google Scholar] [CrossRef] [PubMed]
- European Commission; Joint Research Centre; Directorate-General for Research and Innovation; Institute for Reference Materials and Measurements; Kristiansen, N.; Linsinger, T.; Beloufa, N. The Certification of the Total Glucosinolate and Sulphur Contents of Three Rapeseed (Colza) Materials—BCR-190R, -366R and -367R; Publications Office: Luxembourg, 2001. [Google Scholar]
- Hebert, M.; Serra, E.; Vorobiev, E.; Mhemdi, H. Isolation and Purification of Mustard Glucosinolates by Macroporous Anion-Exchange Resin: Process Optimization and Kinetics’ Modelling. Processes 2022, 10, 191. [Google Scholar] [CrossRef]
- Frank, N.; Dubois, M.; Goldmann, T.; Tarres, A.; Schuster, E.; Robert, F. Semiquantitative Analysis of 3-Butenyl Isothiocyanate To Monitor an Off-flavor in Mustard Seeds and Glycosinolates Screening for Origin Identification. J. Agric. Food Chem. 2010, 58, 3700–3707. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cartridges | Oasis MAX (A) | Bond Elut NH2 (B) | Strata-X-AW (C) | Oasis WAX (D) | Bond Elut DEA (E) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sample load | A1 | B1 | C1 | D1 | E1 | |||||
| Wash 1 | A2 | B2 | √ | C2 | √ | D2 | E2 | |||
| Wash 2 | A3 | C3 | √ | D3 | ||||||
| Elution | A4 | B3 | √ | C4 | √ | D4 | √ | E3 | √ | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Zhang, M.; Pehrsson, P.; Harnly, J.M.; Chen, P.; Sun, J. A Fast and Simple Solid Phase Extraction-Based Method for Glucosinolate Determination: An Alternative to the ISO-9167 Method. Foods 2024, 13, 650. https://doi.org/10.3390/foods13050650
Li Y, Zhang M, Pehrsson P, Harnly JM, Chen P, Sun J. A Fast and Simple Solid Phase Extraction-Based Method for Glucosinolate Determination: An Alternative to the ISO-9167 Method. Foods. 2024; 13(5):650. https://doi.org/10.3390/foods13050650
Chicago/Turabian StyleLi, Yanfang, Mengliang Zhang, Pamela Pehrsson, James M. Harnly, Pei Chen, and Jianghao Sun. 2024. "A Fast and Simple Solid Phase Extraction-Based Method for Glucosinolate Determination: An Alternative to the ISO-9167 Method" Foods 13, no. 5: 650. https://doi.org/10.3390/foods13050650