
There’s Something in What We Eat: An Overview on the Extraction Techniques and Chromatographic Analysis for PFAS Identification in Agri-Food Products




Abstract
1. Introduction
2. Toxicology and Risk Assessment of PFASs
3. Regulation
4. PFAS Analysis on Food Matrices
4.1. Meat

{kind=link}
{kind=link}
| Matrix | Extraction and Pretreatment | Analysis | Recoveries | LOD | LOQ | Ref. |
|---|---|---|---|---|---|---|
| Pork | SPE-WAX for PFAS extraction QuEChERS method for PBDE extraction | LC-HRMS for PFAS analysis, GC-MS/MS for PBDE analysis | 80–119 for PFAS, 88–93 for PBDE | 0.005–0.050 for PFAS | 0.015–0.150 for PFAS | [64] |
| Bovine and swine muscle, bovine liver | QuEChERS method | LC-MS/MS | 80–120 | 0.00826–0.03401 for liver, 0.00592–0.01907 for muscle | 0.050 except for GenX and C6O4 with 0.100 | [70] |
| Chicken nuggets, beef steak, ground beef, chicken leg, pork bacon, pork chop, pork sausage, chicken breast, cured ham, sausage/salami combination, frankfurter (beef/pork), lamb chop, turkey breast, and ground turkey | QuEChERS method | LC-MS/MS | 40–120 | 0.086 PFOS in ground turkey | N/A | [72] |
| Beef, pork | DI-SPME with F-BNN-coated fiber | HPLC-MS/MS | 77.7–110.5 | 0.0036–0.0158 a | N/A | [73] |
| Chicken nuggets | Oasis SPE-WAX cleanup and extraction with ACN solvent | LC-MS/MS | 42–221 | 0.006 for perfluorodecane sulfonate (L-PFDS), 1.76 for perfluorohexyl ethanoic acid (FHEA) | 0.018 for L-PFDS, 5.28 for FHEA | [40] |
| Chicken meat, beef | Alkaline digestion, SLE with ACN solvent followed by SPE-WAX cleanup | LC-MS/MS | 77–89 | 0.0011, 0.0014 and 0.0016 for PFHxS, PFOA and L-PFOS | 0.0031, 0.0034 and 0.0049 for PFHxS, PFOA and L-PFOS | [69] |
| Cow meat (heart, kidney, spleen, liver); sheep meat (heart, kidney, spleen, liver); chicken meat (liver, heart, gizzard) | Extraction with MTBE solvent | LC-MS/MS | 90.6–101.2 for PFOA and 89.2–98.4 for PFOS | 0.038 for PFOA and 0.002 for PFOS | 0.125 for PFOA and 0.007 for PFOS | [74] |
| Beef, chicken, pork | QuEChERSER method | Ultra-performance liquid chromatography–high resolution mass spectrometry (UPLC-HRMS), ultra-high performance liquid chromatography–tandem mass spectrometry (UHPLC-MS/MS) with QqQ | 88 ± 10 | HRMS Method: Beef (0.0009–0.267), chicken (0.0006–0.14879), pork (0.0046–0.3026) QqQ Method: Beef (0.006–0.048), chicken (0.003–0.078), pork (0.020–0.228) | HRMS Method: Beef (0.0027–0.8091), chicken (0.0018–0.4506), pork (0.014–0.9169) QqQ Method: Beef (0.018–0.145), chicken (0.009–0.237), pork (0.061–0.689) | [67] |
Overall Summary
4.2. Milk and Dairy Products
| Matrix | Extraction and Pretreatment | Analysis | Recoveries | LOD | LOQ | Ref. |
|---|---|---|---|---|---|---|
| Breastmilk, retail dairy milk, and infant formulas | QuEChERS method | UHPLC-MS/MS | 65–136 | 0.03–0.05 a 0.04–0.1 b | 0.005–0.050 | [39] |
| Human breastmilk | Extraction with KOH/MeOH solution followed by two SPE purification with WAX SPE and ENVI-Carb SPE cartridges | UHPSFC-MS/MS | N/A | N/A | <0.2 except for perfluorodecyl ethanoic acid (FDEA) (1 ng g−1) | [24] |
| Milk | Extraction with Pb(OAc)2 solution, MeOH and FA followed by SPE cleanup with Strata-X-AW cartridge | UHPLC-MS/MS | N/A | 0.0025–0.75 | 0.005–0.1 | [75] |
| Cow milk | QuEChERS method | LC-MS/MS | 91.3–121.8 | 0.00778–0.01635 | 0.05 for all compounds except for GenX and C6O4 (0.1 ng g−1) | [70] |
| Milk | QuEChERS method | LC-MS/MS | 40–120 | 0.007–0.042 c | N/A | [72] |
| Raw and processed milk | SLE with FA in MeOH and AH in MeOH followed by SPE cleanup with ENVI-Carb and Oasis WAX cartridges | HPLC-MS/MS | 70–141 | 0.8–22 d for all PFAS except 144 d for PFBA | N/A | [79] |
| Ice cream | Ultrasonic-assisted extraction (UAE) with ACN solvent followed by SPE cleanup with Oasis WAX cartridge | UHPLC-Orbitrap HRMS | 52–107 | 0.001–0.009 | 0.002–0.020 | [69] |
| Raw milk and bagged milk | DI-SPME with F-BNN-coated fiber | HPLC-MS/MS | 85–110 | 0.9–3.9 c,d | N/A | [73] |
| Dairy milk and infant formula | QuEChERS method | UHPLC-MS/MS | 93–120 | 5–50 d | 5–50 d | [81] |
| Cow milk and butter | Alkaline digestion, SLE with ACN solvent followed by SPE-WAX cleanup | LC-MS/MS | 93–101 | 0.0011, 0.0014 and 0.0016 for PFHxS, PFOA, and L-PFOS | 0.0031, 0.0034 and 0.0049 for PFHxS, PFOA, and L-PFOS | [69] |
| Breastmilk | SALLE | UHPLC-MS/MS | N/A | 0.66–0.86 d | 2.19–2.87 d | [85] |
| Milk | Extraction with MTBE solvent | LC-MS/MS | 90.6–101.2 for PFOA and 89.2–98.4 for PFOS | 0.038 for PFOA and 0.002 for PFOS | 0.125 for PFOA and 0.007 for PFOS | [74] |
| Milk, cottage cheese, natural yoghurt, kefir (bonny clabber), sour cream, Camembert-type cheese, and butter | QuEChERS method | Micro-HPLC-MS/MS) | 70–120 | 0.003–0.009 | 0.010–0.027 | [77] |
| Retail milk and yogurt | UE-MeOH followed by SPE purification with HLB cartridge | HPLC-MS/MS | 85.4–90.1 for PFOA and 80.3–84.3 for PFOS | 5–10 d for PFOA and PFOS | 15–30 d for PFOA and PFOS | [78] |
| Cow milk | QuEChERS method | LC-MS/MS | 78.5–111 for PFCAs and 72.8–105 for PFSAs | 0.0030 and 0.010 d PFCAs and PFSAs | 0.010 and 0.050 d PFCAs and PFSAs | [82] |
Overall Summary
4.3. Fruit and Vegetables
Overall Summary
4.4. Eggs
Overall Summary
4.5. Fish and Shellfish
Overall Summary
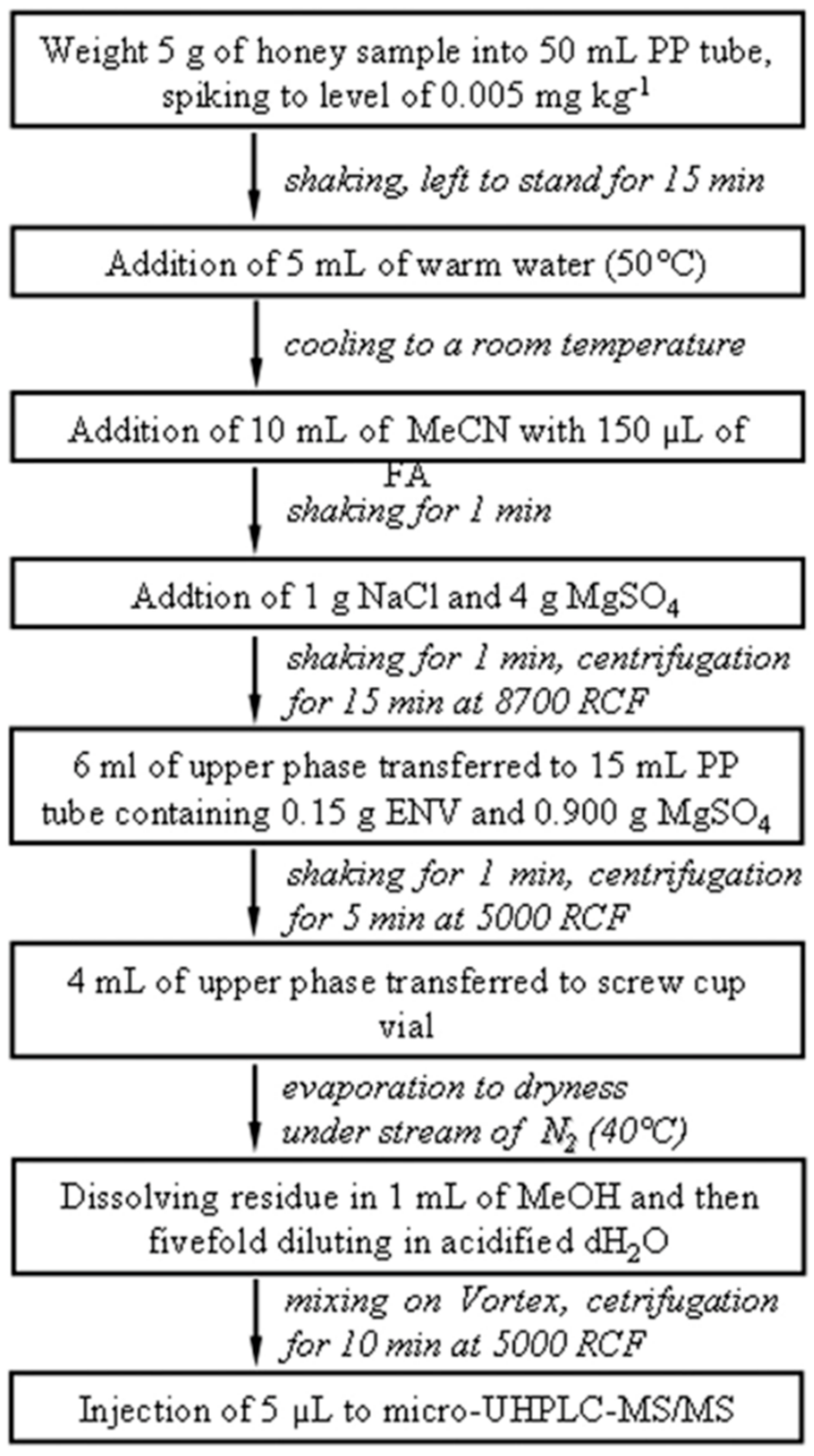
4.6. Honey
4.7. Beverages
Overall Summary
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gravesen, C.R.; Lee, L.S.; Choi, Y.J.; Silveira, M.L.; Judy, J.D. PFAS Release from Wastewater Residuals as a Function of Composition and Production Practices. Environ. Pollut. 2023, 322, 121167. [Google Scholar] [CrossRef]
- An, X.; Lei, H.; Lu, Y.; Xie, X.; Wang, P.; Liao, J.; Liang, Z.; Sun, B.; Wu, Z. Per- and Polyfluoroalkyl Substances (PFASs) in Water and Sediment from a Temperate Watershed in China: Occurrence, Sources, and Ecological Risks. Sci. Total Environ. 2023, 890, 164207. [Google Scholar] [CrossRef]
- Moneta, B.G.; Feo, M.L.; Torre, M.; Tratzi, P.; Aita, S.E.; Montone, C.M.; Taglioni, E.; Mosca, S.; Balducci, C.; Cerasa, M.; et al. Occurrence of Per- and Polyfluorinated Alkyl Substances in Wastewater Treatment Plants in Northern Italy. Sci. Total Environ. 2023, 894, 165089. [Google Scholar] [CrossRef]
- Cara, B.; Lies, T.; Thimo, G.; Robin, L.; Lieven, B. Bioaccumulation and Trophic Transfer of Perfluorinated Alkyl Substances (PFAS) in Marine Biota from the Belgian North Sea: Distribution and Human Health Risk Implications. Environ. Pollut. 2022, 311, 119907. [Google Scholar] [CrossRef]
- Brusseau, M.L.; Anderson, R.H.; Guo, B. PFAS Concentrations in Soils: Background Levels versus Contaminated Sites. Sci. Total Environ. 2020, 740, 140017. [Google Scholar] [CrossRef]
- Cioni, L.; Nikiforov, V.; Coêlho, A.C.M.F.; Sandanger, T.M.; Herzke, D. Total Oxidizable Precursors Assay for PFAS in Human Serum. Environ. Int. 2022, 170, 107656. [Google Scholar] [CrossRef]
- Tian, Z.; Kim, S.-K.; Shoeib, M.; Oh, J.-E.; Park, J.-E. Human Exposure to Per- and Polyfluoroalkyl Substances (PFASs) via House Dust in Korea: Implication to Exposure Pathway. Sci. Total Environ. 2016, 553, 266–275. [Google Scholar] [CrossRef]
- Bil, W.; Govarts, E.; Zeilmaker, M.J.; Woutersen, M.; Bessems, J.; Ma, Y.; Thomsen, C.; Haug, L.S.; Lignell, S.; Gyllenhammar, I.; et al. Approaches to Mixture Risk Assessment of PFASs in the European Population Based on Human Hazard and Biomonitoring Data. Int. J. Hyg. Environ. Health 2023, 247, 114071. [Google Scholar] [CrossRef]
- Chen, X.; Feng, X.; Sun, X.; Li, Y.; Yang, Y.; Shan, G.; Zhu, L. Quantifying Indirect Contribution from Precursors to Human Body Burden of Legacy PFASs Based on Paired Blood and One-Week Duplicate Diet. Environ. Sci. Technol. 2022, 56, 5632–5640. [Google Scholar] [CrossRef]
- Dunder, L.; Lind, P.M.; Salihovic, S.; Stubleski, J.; Kärrman, A.; Lind, L. Changes in Plasma Levels of Per- and Polyfluoroalkyl Substances (PFAS) Are Associated with Changes in Plasma Lipids—A Longitudinal Study over 10 Years. Environ. Res. 2022, 211, 112903. [Google Scholar] [CrossRef]
- Göckener, B.; Weber, T.; Rüdel, H.; Bücking, M.; Kolossa-Gehring, M. Human Biomonitoring of Per- and Polyfluoroalkyl Substances in German Blood Plasma Samples from 1982 to 2019. Environ. Int. 2020, 145, 106123. [Google Scholar] [CrossRef]
- Hall, S.M.; Zhang, S.; Tait, G.H.; Hoffman, K.; Collier, D.N.; Hoppin, J.A.; Stapleton, H.M. PFAS Levels in Paired Drinking Water and Serum Samples Collected from an Exposed Community in Central North Carolina. Sci. Total Environ. 2023, 895, 165091. [Google Scholar] [CrossRef]
- Liu, D.; Tang, B.; Nie, S.; Zhao, N.; He, L.; Cui, J.; Mao, W.; Jin, H. Distribution of Per- and Poly-Fluoroalkyl Substances and Their Precursors in Human Blood. J. Hazard. Mater. 2023, 441, 129908. [Google Scholar] [CrossRef]
- Poothong, S.; Papadopoulou, E.; Padilla-Sánchez, J.A.; Thomsen, C.; Haug, L.S. Multiple Pathways of Human Exposure to Poly- and Perfluoroalkyl Substances (PFASs): From External Exposure to Human Blood. Environ. Int. 2020, 134, 105244. [Google Scholar] [CrossRef]
- Van Beijsterveldt, I.A.L.P.; Van Zelst, B.D.; Van Den Berg, S.A.A.; De Fluiter, K.S.; Van Der Steen, M.; Hokken-Koelega, A.C.S. Longitudinal Poly- and Perfluoroalkyl Substances (PFAS) Levels in Dutch Infants. Environ. Int. 2022, 160, 107068. [Google Scholar] [CrossRef]
- Lu, Y.; Meng, L.; Ma, D.; Cao, H.; Liang, Y.; Liu, H.; Wang, Y.; Jiang, G. The Occurrence of PFAS in Human Placenta and Their Binding Abilities to Human Serum Albumin and Organic Anion Transporter 4. Environ. Pollut. 2021, 273, 116460. [Google Scholar] [CrossRef]
- Gao, K.; Zhuang, T.; Liu, X.; Fu, J.; Zhang, J.; Fu, J.; Wang, L.; Zhang, A.; Liang, Y.; Song, M.; et al. Prenatal Exposure to Per- and Polyfluoroalkyl Substances (PFASs) and Association between the Placental Transfer Efficiencies and Dissociation Constant of Serum Proteins–PFAS Complexes. Environ. Sci. Technol. 2019, 53, 6529–6538. [Google Scholar] [CrossRef]
- Tian, Y.; Zhou, Y.; Miao, M.; Wang, Z.; Yuan, W.; Liu, X.; Wang, X.; Wang, Z.; Wen, S.; Liang, H. Determinants of Plasma Concentrations of Perfluoroalkyl and Polyfluoroalkyl Substances in Pregnant Women from a Birth Cohort in Shanghai, China. Environ. Int. 2018, 119, 165–173. [Google Scholar] [CrossRef]
- Wang, Y.; Han, W.; Wang, C.; Zhou, Y.; Shi, R.; Bonefeld-Jørgensen, E.C.; Yao, Q.; Yuan, T.; Gao, Y.; Zhang, J.; et al. Efficiency of Maternal-Fetal Transfer of Perfluoroalkyl and Polyfluoroalkyl Substances. Environ. Sci. Pollut. Res. 2019, 26, 2691–2698. [Google Scholar] [CrossRef]
- Schildroth, S.; Rodgers, K.M.; Strynar, M.; McCord, J.; Poma, G.; Covaci, A.; Dodson, R.E. Per-and Polyfluoroalkyl Substances (PFAS) and Persistent Chemical Mixtures in Dust from U.S. Colleges. Environ. Res. 2022, 206, 112530. [Google Scholar] [CrossRef]
- Besis, A.; Botsaropoulou, E.; Samara, C.; Katsoyiannis, A.; Hanssen, L.; Huber, S. Perfluoroalkyl Substances (PFASs) in Air-Conditioner Filter Dust of Indoor Microenvironments in Greece: Implications for Exposure. Ecotoxicol. Environ. Saf. 2019, 183, 109559. [Google Scholar] [CrossRef]
- De La Torre, A.; Navarro, I.; Sanz, P.; Mártinez, M.D.L.Á. Occurrence and Human Exposure Assessment of Perfluorinated Substances in House Dust from Three European Countries. Sci. Total Environ. 2019, 685, 308–314. [Google Scholar] [CrossRef]
- Wang, B.; Yao, Y.; Wang, Y.; Chen, H.; Sun, H. Per- and Polyfluoroalkyl Substances in Outdoor and Indoor Dust from Mainland China: Contributions of Unknown Precursors and Implications for Human Exposure. Environ. Sci. Technol. 2022, 56, 6036–6045. [Google Scholar] [CrossRef]
- Moya, J.; Phillips, L. A review of soiul and dust ingestion studies for children. J. Expo. Sci. Environ. Epidemiol. 2014, 24, 545–554. [Google Scholar] [CrossRef]
- Rüdel, H.; Radermacher, G.; Fliedner, A.; Lohmann, N.; Koschorreck, J.; Duffek, A. Tissue Concentrations of Per- and Polyfluoroalkyl Substances (PFAS) in German Freshwater Fish: Derivation of Fillet-to-Whole Fish Conversion Factors and Assessment of Potential Risks. Chemosphere 2022, 292, 133483. [Google Scholar] [CrossRef]
- Semerád, J.; Horká, P.; Filipová, A.; Kukla, J.; Holubová, K.; Musilová, Z.; Jandová, K.; Frouz, J.; Cajthaml, T. The Driving Factors of Per- and Polyfluorinated Alkyl Substance (PFAS) Accumulation in Selected Fish Species: The Influence of Position in River Continuum, Fish Feed Composition, and Pollutant Properties. Sci. Total Environ. 2022, 816, 151662. [Google Scholar] [CrossRef]
- Chiumiento, F.; Bellocci, M.; Ceci, R.; D’Antonio, S.; De Benedictis, A.; Leva, M.; Pirito, L.; Rosato, R.; Scarpone, R.; Scortichini, G.; et al. A New Method for Determining PFASs by UHPLC-HRMS (Q-Orbitrap): Application to PFAS Analysis of Organic and Conventional Eggs Sold in Italy. Food Chem. 2023, 401, 134135. [Google Scholar] [CrossRef]
- Meng, P.; DeStefano, N.J.; Knappe, D.R.U. Extraction and Matrix Cleanup Method for Analyzing Novel Per- and Polyfluoroalkyl Ether Acids and Other Per- and Polyfluoroalkyl Substances in Fruits and Vegetables. J. Agric. Food Chem. 2022, 70, 4792–4804. [Google Scholar] [CrossRef]
- Eick, S.M.; Goin, D.E.; Trowbridge, J.; Cushing, L.; Smith, S.C.; Park, J.-S.; DeMicco, E.; Padula, A.M.; Woodruff, T.J.; Morello-Frosch, R. Dietary Predictors of Prenatal Per- and Poly-Fluoroalkyl Substances Exposure. J. Expo. Sci. Environ. Epidemiol. 2023, 33, 32–39. [Google Scholar] [CrossRef]
- Fiedler, H.; Sadia, M.; Baabish, A.; Sobhanei, S. Perfluoroalkane Substances in National Samples from Global Monitoring Plan Projects (2017–2019). Chemosphere 2022, 307, 136038. [Google Scholar] [CrossRef]
- Bedi, M.; Sapozhnikova, Y.; Taylor, R.B.; Ng, C. Per- and Polyfluoroalkyl Substances (PFAS) Measured in Seafood from a Cross-Section of Retail Stores in the United States. J. Hazard. Mater. 2023, 459, 132062. [Google Scholar] [CrossRef]
- George, S.E.; Baker, T.R.; Baker, B.B. Nonlethal Detection of PFAS Bioaccumulation and Biomagnification within Fishes in an Urban- and Wastewater-Dominant Great Lakes Watershed. Environ. Pollut. 2023, 321, 121123. [Google Scholar] [CrossRef]
- Wu, S.; Yuan, T.; Fu, W.; Dong, H.; Zhang, Y.; Zhang, M.; Jiang, C.; Xu, Q.; Zhang, L.; Qiang, Z. Perfluorinated Compound Correlation between Human Serum and Drinking Water: Is Drinking Water a Significant Contributor? Sci. Total Environ. 2023, 873, 162471. [Google Scholar] [CrossRef]
- Directive (EU) 2020/2184 of the European Parliament and of the Council of 16 December 2020 on the quality of water intended for human consumption. Off. J. Eur. Union 2020, L-435, 1–62. Available online: https://eur-lex.europa.eu/eli/dir/2020/2184/oj (accessed on 12 January 2021).
- Sadia, M.; Nollen, I.; Helmus, R.; Ter Laak, T.L.; Béen, F.; Praetorius, A.; Van Wezel, A.P. Occurrence, Fate, and Related Health Risks of PFAS in Raw and Produced Drinking Water. Environ. Sci. Technol. 2023, 57, 3062–3074. [Google Scholar] [CrossRef]
- Coggan, T.L.; Anumol, T.; Pyke, J.; Shimeta, J.; Clarke, B.O. A Single Analytical Method for the Determination of 53 Legacy and Emerging Per- and Polyfluoroalkyl Substances (PFAS) in Aqueous Matrices. Anal. Bioanal. Chem. 2019, 411, 3507–3520. [Google Scholar] [CrossRef]
- Gao, Y.; Li, X.; Li, X.; Zhang, Q.; Li, H. Simultaneous Determination of 21 Trace Perfluoroalkyl Substances in Fish by Isotope Dilution Ultrahigh Performance Liquid Chromatography Tandem Mass Spectrometry. J. Chromatogr. B 2018, 1084, 45–52. [Google Scholar] [CrossRef]
- Roberts, J.; McNaughtan, M.; De Las Heras Prieto, H. Unwanted Ingredients—Highly Specific and Sensitive Method for the Extraction and Quantification of PFAS in Everyday Foods. Food Anal. Methods 2023, 16, 857–866. [Google Scholar] [CrossRef]
- Abafe, O.A.; Macheka, L.R.; Olowoyo, J.O. Confirmatory Analysis of Per and Polyfluoroalkyl Substances in Milk and Infant Formula Using UHPLC–MS/MS. Molecules 2021, 26, 3664. [Google Scholar] [CrossRef]
- Rawn, D.F.K.; Ménard, C.; Feng, S.Y. Method Development and Evaluation for the Determination of Perfluoroalkyl and Polyfluoroalkyl Substances in Multiple Food Matrices. Food Addit. Contam. Part A 2022, 39, 752–776. [Google Scholar] [CrossRef]
- Jala, A.; Adye, D.R.; Borkar, R.M. Occurrence and Risk Assessments of Per- and Polyfluoroalkyl Substances in Tea Bags from India. Food Control 2023, 151, 109812. [Google Scholar] [CrossRef]
- Solan, M.E.; Koperski, C.P.; Senthilkumar, S.; Lavado, R. Short-Chain per- and Polyfluoralkyl Substances (PFAS) Effects on Oxidative Stress Biomarkers in Human Liver, Kidney, Muscle, and Microglia Cell Lines. Environ. Res. 2023, 223, 115424. [Google Scholar] [CrossRef]
- Liu, S.; Yang, R.; Yin, N.; Wang, Y.-L.; Faiola, F. Environmental and Human Relevant PFOS and PFOA Doses Alter Human Mesenchymal Stem Cell Self-Renewal, Adipogenesis and Osteogenesis. Ecotoxicol. Environ. Saf. 2019, 169, 564–572. [Google Scholar] [CrossRef]
- Di Nisio, A.; Pannella, M.; Vogiatzis, S.; Sut, S.; Dall’Acqua, S.; Rocca, M.S.; Antonini, A.; Porzionato, A.; De Caro, R.; Bortolozzi, M.; et al. Impairment of Human Dopaminergic Neurons at Different Developmental Stages by Perfluoro-Octanoic Acid (PFOA) and Differential Human Brain Areas Accumulation of Perfluoroalkyl Chemicals. Environ. Int. 2022, 158, 106982. [Google Scholar] [CrossRef]
- Hassan, H.F.; Bou Ghanem, H.; Abi Kharma, J.; Abiad, M.G.; Elaridi, J.; Bassil, M. Perfluorooctanoic Acid and Perfluorooctane Sulfonate in Human Milk: First Survey from Lebanon. Int. J. Environ. Res. Public Health 2023, 20, 821. [Google Scholar] [CrossRef]
- Rosenfeld, P.E.; Spaeth, K.R.; Remy, L.L.; Byers, V.; Muerth, S.A.; Hallman, R.C.; Summers-Evans, J.; Barker, S. Perfluoroalkyl Substances Exposure in Firefighters: Sources and Implications. Environ. Res. 2023, 220, 115164. [Google Scholar] [CrossRef]
- California Office of Environmental Health Hazard Assessment. Evidence on the Carcinogenity of Perfluorooctane Sulfonic acid (PFOS) and Its Salts and Transformation and Degradation Precursors. September 2021. Available online: https://oehha.ca.gov/media/downloads/crnr/pfoshid092421.pdf (accessed on 24 December 2021).
- DeWitt, J.C.; Blossom, S.J.; Schaider, L.A. Exposure to Per-Fluoroalkyl and Polyfluoroalkyl Substances Leads to Immunotoxicity: Epidemiological and Toxicological Evidence. J. Expo. Sci. Environ. Epidemiol. 2019, 29, 148–156. [Google Scholar] [CrossRef]
- Agency for Toxic Substances and Disease Registry. Toxicological Profile for Perfluoroalkyls; Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2021. Available online: https://www.atsdr.cdc.gov/toxprofiles/tp200.pdf (accessed on 15 May 2021).
- Porter, A.K.; Kleinschmidt, S.E.; Andres, K.L.; Reusch, C.N.; Krisko, R.M.; Taiwo, O.A.; Olsen, G.W.; Longnecker, M.P. Antibody Response to COVID-19 Vaccines among Workers with a Wide Range of Exposure to per- and Polyfluoroalkyl Substances. Environ. Int. 2022, 169, 107537. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain (EFSA CONTAM Panel); Schrenk, D.; Bignami, M.; Bodin, L.; Chipman, J.K.; del Mazo, J.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L.R.; Leblanc, J.; et al. Risk to Human Health Related to the Presence of Perfluoroalkyl Substances in Food. EFS2 2020, 18, e06223. [Google Scholar] [CrossRef]
- Barbo, N.; Stoiber, T.; Naidenko, O.V.; Andrews, D.Q. Locally Caught Freshwater Fish across the United States Are Likely a Significant Source of Exposure to PFOS and Other Perfluorinated Compounds. Environ. Res. 2023, 220, 115165. [Google Scholar] [CrossRef]
- Ebrahimi, F.; Lewis, A.J.; Sales, C.M.; Suri, R.; McKenzie, E.R. Linking PFAS Partitioning Behavior in Sewage Solids to the Solid Characteristics, Solution Chemistry, and Treatment Processes. Chemosphere 2021, 271, 129530. [Google Scholar] [CrossRef]
- Van Der Veen, I.; Fiedler, H.; De Boer, J. Assessment of the Per- and Polyfluoroalkyl Substances Analysis under the Stockholm Convention—2018/2019. Chemosphere 2023, 313, 137549. [Google Scholar] [CrossRef]
- Kancharla, S.; Choudhary, A.; Davis, R.T.; Dong, D.; Bedrov, D.; Tsianou, M.; Alexandridis, P. GenX in Water: Interactions and Self-Assembly. J. Hazard. Mater. 2022, 428, 128137. [Google Scholar] [CrossRef]
- Drage, D.S.; Sharkey, M.; Berresheim, H.; Coggins, M.; Harrad, S. Rapid Determination of Selected PFAS in Textiles Entering the Waste Stream. Toxics 2023, 11, 55. [Google Scholar] [CrossRef]
- Zacs, D.; Fedorenko, D.; Pasecnaja, E.; Bartkevics, V. Application of Nano-LC—Nano-ESIOrbitrap-MS for Trace Determination of Four Priority PFAS in Food Products Considering Recently Established Tolerable Weekly Intake (TWI) Limits. Anal. Chim. Acta 2023, 1251, 341027. [Google Scholar] [CrossRef]
- Commission Regulation (EU). Regulation 2022/2388 of 7 December 2022 amending Regulation (EC) No 1881/2006 as regards maximum levels of perfluoroalkyl substances in certain foodstuffs. Off. J. Eur. Union 2022, L316, 38–41. Available online: https://eur-lex.europa.eu/eli/reg/2022/2388/oj (accessed on 8 December 2022).
- Delatour, T.; Theurillat, X.; Mujahid, C.; Eriksen, B.; Mottier, P. Determination of Poly- and Perfluoroalkylated Substances in Food: How Consistent Are Results across Laboratories? J. Agric. Food Chem. 2023, 71, 4767–4768. [Google Scholar] [CrossRef]
- EC (European Commission). EU Action Plan: “Toward Zero Pollution for Air, Water and Soil”. Brussels. 2021. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:52021DC0400 (accessed on 15 May 2021).
- Govarts, E.; Gilles, L.; Rodriguez Martin, L.; Santonen, T.; Apel, P.; Alvito, P.; Anastasi, E.; Andersen, H.R.; Andersson, A.-M.; Andryskova, L.; et al. Harmonized Human Biomonitoring in European Children, Teenagers and Adults: EU-Wide Exposure Data of 11 Chemical Substance Groups from the HBM4EU Aligned Studies (2014–2021). Int. J. Hyg. Environ. Health 2023, 249, 114119. [Google Scholar] [CrossRef]
- Ougier, E.; Ganzleben, C.; Lecoq, P.; Bessems, J.; David, M.; Schoeters, G.; Lange, R.; Meslin, M.; Uhl, M.; Kolossa-Gehring, M.; et al. Chemical Prioritisation Strategy in the European Human Biomonitoring Initiative (HBM4EU)—Development and Results. Int. J. Hyg. Environ. Health 2021, 236, 113778. [Google Scholar] [CrossRef]
- Lobo Vicente, J.; Ganzleben, C.; Gasol, R.; Marnane, I.; Gilles, L.; Buekers, J.; Bessems, J.; Colles, A.; Gerofke, A.; David, M.; et al. HBM4EU Results Support the Chemicals’ Strategy for Sustainability and the Zero-Pollution Action Plan. Int. J. Hyg. Environ. Health 2023, 248, 114111. [Google Scholar] [CrossRef]
- Chiesa, L.M.; Lin, S.-K.; Ceriani, F.; Panseri, S.; Arioli, F. Levels and Distribution of PBDEs and PFASs in Pork from Different European Countries. Food Addit. Contam. Part A 2018, 35, 2414–2423. [Google Scholar] [CrossRef]
- Giromini, C.; Givens, D.I. Benefits and Risks Associated with Meat Consumption during Key Life Processes and in Relation to the Risk of Chronic Diseases. Foods 2022, 11, 2063. [Google Scholar] [CrossRef]
- Lin, Y.; Liu, R.; Hu, F.; Liu, R.; Ruan, T.; Jiang, G. Simultaneous Qualitative and Quantitative Analysis of Fluoroalkyl Sulfonates in Riverine Water by Liquid Chromatography Coupled with Orbitrap High Resolution Mass Spectrometry. J. Chromatogr. A 2016, 1435, 66–74. [Google Scholar] [CrossRef]
- Taylor, R.B.; Sapozhnikova, Y. Comparison and Validation of the QuEChERSER Mega-Method for Determination of per- and Polyfluoroalkyl Substances in Foods by Liquid Chromatography with High-Resolution and Triple Quadrupole Mass Spectrometry. Anal. Chim. Acta 2022, 1230, 340400. [Google Scholar] [CrossRef]
- Zhou, Y.; Lian, Y.; Sun, X.; Fu, L.; Duan, S.; Shang, C.; Jia, X.; Wu, Y.; Wang, M. Determination of 20 Perfluoroalkyl Substances in Greenhouse Vegetables with a Modified One-Step Pretreatment Approach Coupled with Ultra Performance Liquid Chromatography Tandem Mass Spectrometry(UPLC-MS-MS). Chemosphere 2019, 227, 470–479. [Google Scholar] [CrossRef]
- Sadia, M.; Yeung, L.W.Y.; Fiedler, H. Trace Level Analyses of Selected Perfluoroalkyl Acids in Food: Method Development and Data Generation. Environ. Pollut. 2020, 263, 113721. [Google Scholar] [CrossRef]
- Gallocchio, F.; Moressa, A.; Zonta, G.; Angeletti, R.; Lega, F. Fast and Sensitive Analysis of Short- and Long-Chain Perfluoroalkyl Substances in Foods of Animal Origin. Molecules 2022, 27, 7899. [Google Scholar] [CrossRef]
- Gallocchio, F.; Mancin, M.; Belluco, S.; Moressa, A.; Angeletti, R.; Lorenzetto, M.; Arcangeli, G.; Ferrè, N.; Ricci, A.; Russo, F. Investigation of Levels of Perfluoroalkyl Substances in Freshwater Fishes Collected in a Contaminated Area of Veneto Region, Italy. Environ. Sci. Pollut. Res. 2022, 29, 20996–21011. [Google Scholar] [CrossRef]
- Genualdi, S.; Young, W.; DeJager, L.; Begley, T. Method Development and Validation of Per- and Polyfluoroalkyl Substances in Foods from FDA’s Total Diet Study Program. J. Agric. Food Chem. 2021, 69, 5599–5606. [Google Scholar] [CrossRef]
- Li, J.; Gao, Y.; Wan, Y.; Liu, J.; Liu, L.; Wang, J.; Sun, X.; Pi, F.; Chen, X. A Novel Analytical Strategy for the Determination of Perfluoroalkyl Acids in Various Food Matrices Using a Home-Made Functionalized Fluorine Interaction SPME in Combination with LC-MS/MS. Food Chem. 2022, 366, 130572. [Google Scholar] [CrossRef]
- Sungur, Ş.; Köroğlu, M.; Turgut, F. Determination of Perfluorooctanoic Acid (PFOA) and Perfluorooctane Sulfonic Acid (PFOS) in Food and Beverages. Int. J. Environ. Anal. Chem. 2018, 98, 360–368. [Google Scholar] [CrossRef]
- Berendsen, B.J.A.; Lakraoui, F.; Leenders, L.; Van Leeuwen, S.P.J. The Analysis of Perfluoroalkyl Substances at Ppt Level in Milk and Egg Using UHPLC-MS/MS. Food Addit. Contam. Part A 2020, 37, 1707–1718. [Google Scholar] [CrossRef]
- Chen, W.-L.; Bai, F.-Y.; Chang, Y.-C.; Chen, P.-C.; Chen, C.-Y. Concentrations of Perfluoroalkyl Substances in Foods and the Dietary Exposure among Taiwan General Population and Pregnant Women. J. Food Drug Anal. 2018, 26, 994–1004. [Google Scholar] [CrossRef]
- Sznajder-Katarzyńska, K.; Surma, M.; Wiczkowski, W.; Cieślik, E. The Perfluoroalkyl Substance (PFAS) Contamination Level in Milk and Milk Products in Poland. Int. Dairy J. 2019, 96, 73–84. [Google Scholar] [CrossRef]
- Xing, Z.; Lu, J.; Liu, Z.; Li, S.; Wang, G.; Wang, X. Occurrence of Perfluorooctanoic Acid and Perfluorooctane Sulfonate in Milk and Yogurt and Their Risk Assessment. Int. J. Environ. Res. Public Health 2016, 13, 1037. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.I.; Becanova, J.; Lohmann, R. A Sensitive Method for the Detection of Legacy and Emerging Per- and Polyfluorinated Alkyl Substances (PFAS) in Dairy Milk. Anal. Bioanal. Chem. 2022, 414, 1235–1243. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, X.; Wang, X.; Deji, Z.; Lee, H.K. Occurrence of Perfluoroalkyl and Polyfluoroalkyl Substances in Ice Cream, Instant Noodles, and Bubble Tea. J. Agric. Food Chem. 2022, 70, 10836–10846. [Google Scholar] [CrossRef]
- Macheka, L.R.; Olowoyo, J.O.; Mugivhisa, L.L.; Abafe, O.A. Determination and Assessment of Human Dietary Intake of per and Polyfluoroalkyl Substances in Retail Dairy Milk and Infant Formula from South Africa. Sci. Total Environ. 2021, 755, 142697. [Google Scholar] [CrossRef]
- Yu, Y.; Xu, D.; Lu, M.; Zhou, S.; Peng, T.; Yue, Z.; Zhou, Y. QuEChERs Combined with Online Interference Trapping LC-MS/MS Method for the Simultaneous Determination of 20 Polyfluoroalkane Substances in Dietary Milk. J. Agric. Food Chem. 2015, 63, 4087–4095. [Google Scholar] [CrossRef] [PubMed]
- Surma, M.; Wiczkowski, W.; Cieślik, E.; Zieliński, H. Method Development for the Determination of PFOA and PFOS in Honey Based on the Dispersive Solid Phase Extraction (d-SPE) with Micro-UHPLC–MS/MS System. Microchem. J. 2015, 121, 150–156. [Google Scholar] [CrossRef]
- Zheng, G.; Schreder, E.; Dempsey, J.C.; Uding, N.; Chu, V.; Andres, G.; Sathyanarayana, S.; Salamova, A. Per- and Polyfluoroalkyl Substances (PFAS) in Breast Milk: Concerning Trends for Current-Use PFAS. Environ. Sci. Technol. 2021, 55, 7510–7520. [Google Scholar] [CrossRef]
- Serrano, L.; Iribarne-Durán, L.M.; Suárez, B.; Artacho-Cordón, F.; Vela-Soria, F.; Peña-Caballero, M.; Hurtado, J.A.; Olea, N.; Fernández, M.F.; Freire, C. Concentrations of Perfluoroalkyl Substances in Donor Breast Milk in Southern Spain and Their Potential Determinants. Int. J. Hyg. Environ. Health 2021, 236, 113796. [Google Scholar] [CrossRef] [PubMed]
- D’Hollander, W.; Herzke, D.; Huber, S.; Hajslova, J.; Pulkrabova, J.; Brambilla, G.; De Filippis, S.P.; Bervoets, L.; De Voogt, P. Occurrence of Perfluorinated Alkylated Substances in Cereals, Salt, Sweets and Fruit Items Collected in Four European Countries. Chemosphere 2015, 129, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Sznajder-Katarzyńska, K.; Surma, M.; Cieślik, E.; Wiczkowski, W. The Perfluoroalkyl Substances (PFASs) Contamination of Fruits and Vegetables. Food Addit. Contam. Part A 2018, 35, 1776–1786. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Yu, W.-J.; Liu, Y.; Wang, X.; Jin, Y.-H.; Dong, G.-H. Perfluoroalkyl Substances in Groundwater and Home-Produced Vegetables and Eggs around a Fluorochemical Industrial Park in China. Ecotoxicol. Environ. Saf. 2019, 171, 199–205. [Google Scholar] [CrossRef]
- Surma, M.; Sadowska-Rociek, A.; Draszanowska, A. Levels of Contamination by Pesticide Residues, Polycyclic Aromatic Hydrocarbons (PAHs), and 5-Hydroxymethylfurfural (HMF) in Honeys Retailed in Europe. Arch. Environ. Contam. Toxicol. 2023, 84, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Scordo, C.V.A.; Checchini, L.; Renai, L.; Orlandini, S.; Bruzzoniti, M.C.; Fibbi, D.; Mandi, L.; Ouazzani, N.; Del Bubba, M. Optimization and Validation of a Method Based on QuEChERS Extraction and Liquid Chromatographic–Tandem Mass Spectrometric Analysis for the Determination of Perfluoroalkyl Acids in Strawberry and Olive Fruits, as Model Crops with Different Matrix Characteristics. J. Chromatogr. A 2020, 1621, 461038. [Google Scholar] [CrossRef] [PubMed]
- Genualdi, S.; Jeong, N.; deJager, L.; Begley, T. Investigation into Perfluoroalkyl Substances (PFASs) in a Cranberry Bog: Method Development and Sampling Results. Food Addit. Contam. Part A 2017, 34, 2181–2189. [Google Scholar] [CrossRef] [PubMed]
- Lasters, R.; Groffen, T.; Eens, M.; Coertjens, D.; Gebbink, W.A.; Hofman, J.; Bervoets, L. Home-Produced Eggs: An Important Human Exposure Pathway of Perfluoroalkylated Substances (PFAS). Chemosphere 2022, 308, 136283. [Google Scholar] [CrossRef] [PubMed]
- Tahziz, A.; Mohamad Haron, D.E.; Aziz, M.Y. Liquid Chromatographic Tandem Mass Spectrometric (LC-MS/MS) Determination of Perfluorooctane Sulfonate (PFOS) and Perfluorooctanoic Acid (PFOA) in the Yolk of Poultry Eggs in Malaysia. Molecules 2020, 25, 2335. [Google Scholar] [CrossRef]
- Zafeiraki, E.; Costopoulou, D.; Vassiliadou, I.; Leondiadis, L.; Dassenakis, E.; Hoogenboom, R.L.A.P.; Van Leeuwen, S.P.J. Perfluoroalkylated Substances (PFASs) in Home and Commercially Produced Chicken Eggs from the Netherlands and Greece. Chemosphere 2016, 144, 2106–2112. [Google Scholar] [CrossRef]
- Gazzotti, T.; Sirri, F.; Ghelli, E.; Zironi, E.; Zampiga, M.; Pagliuca, G. Perfluoroalkyl Contaminants in Eggs from Backyard Chickens Reared in Italy. Food Chem. 2021, 362, 130178. [Google Scholar] [CrossRef] [PubMed]
- Young, W.; Wiggins, S.; Limm, W.; Fisher, C.M.; DeJager, L.; Genualdi, S. Analysis of Per- and Poly(Fluoroalkyl) Substances (PFASs) in Highly Consumed Seafood Products from U.S. Markets. J. Agric. Food Chem. 2022, 70, 13545–13553. [Google Scholar] [CrossRef]
- Chen, M.; Zhu, L.; Wang, Q.; Shan, G. Tissue Distribution and Bioaccumulation of Legacy and Emerging Per-and Polyfluoroalkyl Substances (PFASs) in Edible Fishes from Taihu Lake, China. Environ. Pollut. 2021, 268, 115887. [Google Scholar] [CrossRef]
- Ciccotelli, V.; Abete, M.C.; Squadrone, S. PFOS and PFOA in Cereals and Fish: Development and Validation of a High Performance Liquid Chromatography-Tandem Mass Spectrometry Method. Food Control 2016, 59, 46–52. [Google Scholar] [CrossRef]
- Abafe, O.A.; Macheka, L.R.; Abafe, O.T.; Chokwe, T.B. Concentrations and Human Exposure Assessment of per and Polyfluoroalkyl Substances in Farmed Marine Shellfish in South Africa. Chemosphere 2021, 281, 130985. [Google Scholar] [CrossRef] [PubMed]
- Lertassavakorn, T.; Pholphana, N.; Rangkadilok, N.; Suriyo, T.; Satayavivad, J. Determination of Perfluorooctane Sulphonate and Perfluorooctanoic Acid in Seafood and Water from Map Ta Phut Industrial Estate Area, Thailand. Food Addit. Contam. Part A 2021, 38, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, M.; Buffo, A.; Cappelli, F.; Pascariello, S.; Polesello, S.; Valsecchi, S.; Volta, P.; Bettinetti, R. Perfluoroalkyl Acids in Fish of Italian Deep Lakes: Environmental and Human Risk Assessment. Sci. Total Environ. 2019, 653, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, J.; Flor, M.; Karl, H.; Lahrssen-Wiederholt, M. Perfluoroalkyl Substances (PFAS) in Beaked Redfish (Sebastes Mentella) and Cod (Gadus Morhua) from Arctic Fishing Grounds of Svalbard. Food Addit. Contam. Part B 2020, 13, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Couderc, M.; Poirier, L.; Zalouk-Vergnoux, A.; Kamari, A.; Blanchet-Letrouvé, I.; Marchand, P.; Vénisseau, A.; Veyrand, B.; Mouneyrac, C.; Le Bizec, B. Occurrence of POPs and Other Persistent Organic Contaminants in the European Eel (Anguilla Anguilla) from the Loire Estuary, France. Sci. Total Environ. 2015, 505, 199–215. [Google Scholar] [CrossRef]
- Ruffle, B.; Vedagiri, U.; Bogdan, D.; Maier, M.; Schwach, C.; Murphy-Hagan, C. Perfluoroalkyl Substances in U.S. Market Basket Fish and Shellfish. Environ. Res. 2020, 190, 109932. [Google Scholar] [CrossRef]
- Taylor, M.D.; Johnson, D.D.; Nilsson, S.; Lin, C.-Y.; Braeunig, J.; Mueller, J.; Bowles, K.C. Trial of a Novel Experimental Design to Test Depuration of PFASs from the Edible Tissues of Giant Mud Crab Following Exposure under Natural Conditions in the Wild. Sci. Total Environ. 2021, 758, 143650. [Google Scholar] [CrossRef]
- Taylor, M.D.; Nilsson, S.; Bräunig, J.; Bowles, K.C.; Cole, V.; Moltschaniwskyj, N.A.; Mueller, J.F. Do Conventional Cooking Methods Alter Concentrations of Per- and Polyfluoroalkyl Substances (PFASs) in Seafood? Food Chem. Toxicol. 2019, 127, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Lewiński, R.; Hernik, A.; Liszewska, M.; Buckley, B.; Czaja, K.; Korcz, W.; Słomczyńska, A.; Struciński, P. Validation of a Modified QuEChERS Method for the Determination of Selected Organochlorine Compounds in Honey. Molecules 2023, 28, 842. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, C.; De Cristofaro, A.; Nuzzo, A.; Notardonato, I.; Ganassi, S.; Iafigliola, L.; Sardella, G.; Ciccone, M.; Nugnes, D.; Passarella, S.; et al. Biomonitoring of Polycyclic Aromatic Hydrocarbons, Heavy Metals, and Plasticizers Residues: Role of Bees and Honey as Bioindicators of Environmental Contamination. Environ. Sci. Pollut. Res. 2023, 30, 44234–44250. [Google Scholar] [CrossRef] [PubMed]
- Surma, M.; Zieliński, H.; Piskuła, M. Levels of Contamination by Perfluoroalkyl Substances in Honey from Selected European Countries. Bull. Environ. Contam. Toxicol. 2016, 97, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Olomukoro, A.A.; Emmons, R.V.; Godage, N.H.; Cudjoe, E.; Gionfriddo, E. Ion Exchange Solid Phase Microextraction Coupled to Liquid Chromatography/Laminar Flow Tandem Mass Spectrometry for the Determination of Perfluoroalkyl Substances in Water Samples. J. Chromatogr. A 2021, 1651, 462335. [Google Scholar] [CrossRef]
- Ciofi, L.; Renai, L.; Rossini, D.; Ancillotti, C.; Falai, A.; Fibbi, D.; Bruzzoniti, M.C.; Santana-Rodriguez, J.J.; Orlandini, S.; Del Bubba, M. Applicability of the Direct Injection Liquid Chromatographic Tandem Mass Spectrometric Analytical Approach to the Sub-Ng L−1 Determination of Perfluoro-Alkyl Acids in Waste, Surface, Ground and Drinking Water Samples. Talanta 2018, 176, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Xian, Y.; Liang, M.; Wu, Y.; Wang, B.; Hou, X.; Dong, H.; Wang, L. Fluorine and Nitrogen Functionalized Magnetic Graphene as a Novel Adsorbent for Extraction of Perfluoroalkyl and Polyfluoroalkyl Substances from Water and Functional Beverages Followed by HPLC-Orbitrap HRMS Determination. Sci. Total Environ. 2020, 723, 138103. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y.; Takahashi, M.; Tsutsumi, T.; Inoue, K.; Akiyama, H. Monitoring Analysis of Perfluoroalkyl Substances and F-53B in Bottled Water, Tea and Juice Samples by LC-MS/MS. Chem. Pharm. Bull. 2021, 69, 286–290. [Google Scholar] [CrossRef]
- Ao, J.; Yuan, T.; Xia, H.; Ma, Y.; Shen, Z.; Shi, R.; Tian, Y.; Zhang, J.; Ding, W.; Gao, L.; et al. Characteristic and Human Exposure Risk Assessment of Per- and Polyfluoroalkyl Substances: A Study Based on Indoor Dust and Drinking Water in China. Environ. Pollut. 2019, 254, 112873. [Google Scholar] [CrossRef]
- Borrull, J.; Colom, A.; Fabregas, J.; Pocurull, E.; Borrull, F. A Liquid Chromatography Tandem Mass Spectrometry Method for Determining 18 Per- and Polyfluoroalkyl Substances in Source and Treated Drinking Water. J. Chromatogr. A 2020, 1629, 461485. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.J.; Ojeda, N.; Jacangelo, J.G.; Schwab, K.J. Detection of Ultrashort-Chain and Other per- and Polyfluoroalkyl Substances (PFAS) in U.S. Bottled Water. Water Res. 2021, 201, 117292. [Google Scholar] [CrossRef] [PubMed]
- Ünlü Endirlik, B.; Bakır, E.; Boşgelmez, İ.İ.; Eken, A.; Narin, İ.; Gürbay, A. Assessment of Perfluoroalkyl Substances Levels in Tap and Bottled Water Samples from Turkey. Chemosphere 2019, 235, 1162–1171. [Google Scholar] [CrossRef] [PubMed]
- Harrad, S.; Wemken, N.; Drage, D.S.; Abdallah, M.A.-E.; Coggins, A.-M. Perfluoroalkyl Substances in Drinking Water, Indoor Air and Dust from Ireland: Implications for Human Exposure. Environ. Sci. Technol. 2019, 53, 13449–13457. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Zhang, L.; Huang, Z.; Liu, Y.; Wu, N.; He, J.; Zhang, Z.; Zhang, Y.; Niu, Z. Perfluoroalkyl Acids in Drinking Water of China in 2017: Distribution Characteristics, Influencing Factors and Potential Risks. Environ. Int. 2019, 123, 87–95. [Google Scholar] [CrossRef]

| Matrix | Extraction and Pretreatment | Analysis | Recoveries | LOD | LOQ | Ref. |
|---|---|---|---|---|---|---|
| Home-produced vegetables (tomato, cucumber, eggplant, pepper, Chinese cabbage) | Extraction with MTBE solvent followed by cleanup SPE with Water Oasis WAX cartridge | HPLC-MS/MS | 82–95 | N/A | 0.20 | [88] |
| Berries, citrus fruit, pipe and stone fruit, melons, grape, bananas | Extraction with KOH/MeOH solution followed by SPE cleanup with Oasis WAX cartridge | UPLC-MS/MS | 67–106 | N/A | 0.001–0.04 | [86] |
| Cranberry | QuEChERS method | LC-MS/MS | 69–109 | 0.20–5.6 | N/A | [91] |
| Strawberries, Brussels sprouts pepper, green beans, cabbage, collards, squash, apple, asparagus, avocado, banana, peaches, blueberries, broccoli, cantaloupe, carrot, cauliflower, celery, corn, cucumber, eggplant, lettuce, mushrooms, onion, orange, pear, pineapple, spinach, potato, tomato, watermelon, grapefruit | QuEChERS method | LC-MS/MS | 40–120 | 0.02–0.107 | N/A | [72] |
| Blackberries, blueberries, corn kernels (corn), grapes, okra, peaches, pecans, potatoes, squash, and tomatoes | Extraction with basic MeOH solvent followed by SPE cleanup with Oasis WAX SPE cartridge | LC-QqQ-MS | 50–150 | N/A | 0.025–0.25 a | [28] |
| Lettuce, spinach, and tomato | Oasis SPE-WAX cleanup and extraction with ACN solvent | LC-MS/MS | 47–252 | 0.006–1.76 (L-FDS and FHEA, respectively) | 0.018–5.28 (L-FDS and FHEA, respectively) | [40] |
| Strawberry and olive fruits | QuEChERS method | LC-MS/MS | 65–89 (strawberry) and 75–97 (olive fruits) | 0.0007–0.109 b | 0.0029–0.393 a (strawberry) 0.0026–0.127 a (olive fruits) | [90] |
| Vegetable (parsley, tomato, potato, peas) | Extraction with MTBE solvent | LC-MS/MS | 91–101 (PFOA) and 89–98 (PFOS) | 0.038 (PFOA) and 0.002 (PFOS) | 0.125 (PFOA) and 0.007 (PFOS) | [74] |
| Banana, apple, lemon, orange, cherry and strawberry, potato, beetroot, carrot, white cabbage, and tomato | QuEChERS method | Micro-HPLC-MS/MS | 85–97 | 0.002–0.009 | 0.006–0.024 | [87] |
| Cucumber, lettuce, eggplant, tomato, and leek | QuEChERS method | LC-MS/MS | 55–119 | 0.003–0.034 | N/A | [68] |
| Matrix | Extraction and Pretreatment | Analysis | Recoveries | LOD | LOQ | Ref. |
|---|---|---|---|---|---|---|
| Eggs | Extraction with Pb(OAc)2 solution, MeOH, and FA followed by SPE cleanup with Strata-X-AW cartridge | UHPLC-MS/MS | N/A | 0.025–2.5 | 0.025–5 | [75] |
| Eggs and egg products | Alkaline digestion with a NaOH followed by SPE | UPLC-MS/MS | N/A | 0.25 | 0.10 | [95] |
| Home-produced eggs | Extraction with ACN solvent and cleanup using graphitized Envi-Carb powder cartridge | UPLC-MS/MS | 12–35 (PFCAs) and 25–51 (PFSAs) | 11–100 (PFCASs) a and 0–100 (PFSAs) a | 0.080–0.21 (PFCAs) and 0.13–2.5 (PFSAs) | [92] |
| Eggs | Alkaline digestion, SLE with ACN solvent followed by SPE-WAX cleanup | LC-MS/MS | 76–93 | 0.0011, 0.0014 and 0.0016 for PFHxS, PFOA, and L-PFOS | 0.0031, 0.0034 and 0.0049 for PFHxS, PFOA, and L-PFOS | [69] |
| Eggs | Extraction with MTBE solvent | LC-MS/MS | 91–101 for PFOA and 89–98 for PFOS | 0.038 (PFOA) and 0.002 (PFOS) | 0.125 (PFOA) and 0.007 (PFOS | [74] |
| Yolk and poultry eggs | Simple and rapid protein precipitation extraction with ACN solvent | LC-MS/MS | 93–102 (PFOS) 84–91 (PFOA) | 0.1 (PFOS) and 0.02 (PFOA) | 0.5 (PFOS) b and 0.1 (PFOA) b | [93] |
| Liquid and powdered eggs | QuEChERSER method | UPLC-HRMS, UHPLC-MS/MS QqQ | 89 ± 9 | HRMS Method: Liquid eggs (0.0015–0.0161), powdered eggs (0.0003–0.0369) QqQ Method: Liquid eggs (0.003–0.036), powdered eggs (0.005–0.118) | HRMS Method: Liquid eggs (0.0045–0.0489), powdered eggs (0.0009–0.1119) QqQ Method: Liquid eggs (0.008–0.142), powdered eggs (0.016–0.118) | [67] |
| Home- and commercially produced chicken eggs | Alkaline digestion with NaOH with MeOH as solvent extraction followed by SPE cleanup with Oasis WAX SPE cartridge | LC-MS/MS | 60–115 | 0.15 | 0.5 | [94] |
| Matrix | Extraction and Pretreatment | Analysis | Recoveries | LOD | LOQ | Ref. |
|---|---|---|---|---|---|---|
| Marine Shellfish | Extraction with ACN solvent and water | UHPLC-MS/MS | 77–119 | 0.03–0.1 a 0.05–0.16 b | 0.005–0.05 | [99] |
| Fish | Extraction with MTBE solvent followed by cleanup with GBC-SPE cartridge | UPLC-MS/MS | 53–116 | 0.000200–0.0560 c | N/A | [97] |
| Fish | Extraction with MeOH solvent followed by SPE cleanup with Water Oasis WAX cartridge | LC-MS/MS | 99–102 (PFOA) and 96–108 (PFOS) | 0.20–0.47 | 0.50–0.70 | [98] |
| Muscles and bile of European eel (Anguilla anguilla) | Liquid solid extraction (LSE) with KOH of MeOH followed by purification with SPE WAX cartridge and second cleanup with Envi-carb cartridge | LC-MS/MS | 47.6–92.4 | 0.003–0.46 d | 0.006–1.259 | [103] |
| Fish | QuEChERS method | LC-MS/MS | 67.9–120.2 | 0.00369–0.017.33 | 0.05 except for GenX and C6O4 (0.1 ng g−1) | [70] |
| Salmon, tilapia | QuEChERS method | LC-MS/MS | 40–120 | 0.021–0.09 c | N/A | [72] |
| Beaked redfish and cod | Extraction with MeOH solvent followed by cleanup with Oasis WAX column | HPLC-MS/MS | 90–100 | 0.2–1.0 (liver), 0.05–0.2 (Belly flap and fillet) | 0.3–2.0 (liver), 0.1–0.2 (Belly flap and fillet) | [102] |
| Seafood | Extraction with MeOH solvent followed by SPE cleanup with Oasis WAX SPE cartridge | UHPLC-MS/MS | 94.7–100.8 | N/A | 0.024–0.048 | [100] |
| Fish | SE in acidified water and ACN solution followed by purification with Hybrid-SPE®-Phospholipid Ultra cartridges | UHPLC-MS/MS coupled with TFC | N/A | 0.1–1.2 | 0.03–0.4 | [101] |
| Fish | Oasis SPE-WAX cleanup and extraction with ACN solvent | LC-MS/MS | 57.5–116 | 0.006–1.76 | 0.018–5.28 | [40] |
| Fish and shellfish | Digestion and SPE | UPLC-MS/MS | N/A | 0.412–0.560 and 0.698–0.947 (PFNS) | 0.98–1.3 | [104] |
| Fish | Alkaline digestion, SLE with ACN solvent followed by SPE-WAX cleanup | LC-MS/MS | >70 | 0.0011, 0.0014 and 0.0016 for PFHxS, PFOA and L-PFOS | 0.0031, 0.0034 and 0.0049 for PFHxS, PFOA and L-PFOS | [69] |
| Fish | Extraction with MTBE solvent | LC-MS/MS | 89.2–98.4 (PFOS) and 90.6–101.2 (PFOA) | 0.002–0.007 (PFOS) and 0.038–0.125 (PFOA) | 0.007–0.125 | [74] |
| Crabs | Digestion with NaOH in MeOH and SE with ACN, purification with LLE with C6H14 solvent followed by cleanup through Bond Elut carbon cartridges | HPLC-MS/MS | N/A | 1.2 (PFOS) | N/A | [105] |
| Catfish tissue | QuEChERSER method | UPLC-HRMS UHPLC-MS/MS QqQ | 84 | HRMS Method: 1.4–309.6 QqQ Method: 2–54 | HRMS Method: 4.2–938.3 QqQ Method: Method: 5–163 | [67] |
| Seafood (tuna, salmon, shrimp, tilapia, crab, cod, pollock, clam) | QuEChERS method | LC-MS/MS | 44–160 | <0.1 c 0.345 (PFBA), 207 (PFPeA) | N/A | [96] |
| Matrix | Extraction and Pretreatment | Analysis | Recoveries | LOD | LOQ | Ref. |
|---|---|---|---|---|---|---|
| Honey | QuEChERS method | micro-UHPLC-MS/MS | 40–84 (PFOA) and 7–87 (PFOS) | 0.016 (PFOA) and 0.040 (PFOS) | 0.052 (PFOA) and 0.134 (PFOS) | [83] |
| Honey | QuEChERS method | micro-UHPLC-MS/MS | 75–93 | 0.014–0.040 | 0.042–0.134 | [109] |
| Matrix | Extraction and Pretreatment | Analysis | Recoveries | LOD | LOQ | Ref. |
|---|---|---|---|---|---|---|
| Tap and bottled water | ASE and SPE cleanup with Oasis WAX cartridge | UPLC-MS/MS | 80.2–95.2 | 0.00008–0.00068 a | 0.00026–0.00225 a | [114] |
| Tap and bottled water | Large volume direct injection (LVDI) | LC-MS/MS | 87–114 | 0.05–1.0 b | 0.1–2.0 c | [115] |
| Bottled water | SPE with Oasis HLB and Oasis WAX cartridges | LC-MS/MS | 75–111 | 0.11–1.04 b | N/A | [116] |
| Drinking water | DI | UHPLC-MS/MS | N/A | 0.014–0.44 b | 0.030–1.0 c | [111] |
| Drinking water | Offline SPE with Oasis WAX weak anion exchange cartridges | LC-MS/MS | 66–138 | 0.28–18 b | 0.35–26 c | [36] |
| Tap and bottled water | SPE with Oasis WAX cartridges | UHPLC-MS/MS | 82–105 | 0.07–0.12 b | N/A | [117] |
| Tap water | SPE with Oasis WAX cartridges | HPLC-MS | 80–120 | N/A | N/A | [118] |
| Bubble tea | USAE with ACN solvent followed by SPE cleanup with Oasis WAX cartridge | UHPLC-Orbitrap HRMS | 43–114 | 0.001–0.012 | 0.002–0.057 | [80] |
| Bottled water, juice, and tea | SPE with Presep PFC-II cartridges | LC-MS/MS | 80.4–118.8 | 0.1–0.8 | 0.2–1.6 | [113] |
| Tap water | Extraction with MeOH solvent followed by SPE cleanup with Oasis WAX SPE cartridge | UHPLC-MS/MS | 97.6 (PFOA) and 106.2 (PFOS) | N/A | 0.125–0.25 | [100] |
| Tap water | SPE with Waters Oasis WAX cartridge | HPLC-MS/MS | 60–125 | 0.01–0.13 | 0.01–0.47 | [119] |
| Tap and bottles water | SPME procedure, HLB-WAX/PAN fiber | UHPLC-MS/MS | N/A | N/A | 1–2.5 | [110] |
| Juice | Extraction with MTBE solvent | LC-MS/MS | 90.6–101.2 (PFOA) and 89.2–98.4 (PFOS) | 0.038 a (PFOA) and 0.002 a (PFOS) | 0.125 a (PFOA) and 0.007 a (PFOS) | [74] |
| Drinking water, tap water, and functional beverages (Mizone, vitamin water, and soda water) | MSPE procedure | HPLC-Orbitrap-HRMS | 71.9–117.6 | 3–15 | 10–49 | [82] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iannone, A.; Carriera, F.; Passarella, S.; Fratianni, A.; Avino, P. There’s Something in What We Eat: An Overview on the Extraction Techniques and Chromatographic Analysis for PFAS Identification in Agri-Food Products. Foods 2024, 13, 1085. https://doi.org/10.3390/foods13071085
Iannone A, Carriera F, Passarella S, Fratianni A, Avino P. There’s Something in What We Eat: An Overview on the Extraction Techniques and Chromatographic Analysis for PFAS Identification in Agri-Food Products. Foods. 2024; 13(7):1085. https://doi.org/10.3390/foods13071085
Chicago/Turabian StyleIannone, Alessia, Fabiana Carriera, Sergio Passarella, Alessandra Fratianni, and Pasquale Avino. 2024. "There’s Something in What We Eat: An Overview on the Extraction Techniques and Chromatographic Analysis for PFAS Identification in Agri-Food Products" Foods 13, no. 7: 1085. https://doi.org/10.3390/foods13071085
APA StyleIannone, A., Carriera, F., Passarella, S., Fratianni, A., & Avino, P. (2024). There’s Something in What We Eat: An Overview on the Extraction Techniques and Chromatographic Analysis for PFAS Identification in Agri-Food Products. Foods, 13(7), 1085. https://doi.org/10.3390/foods13071085