
Medium-Chain Triglyceride Oil and Dietary Intervention Improved Body Composition and Metabolic Parameters in Children with Glycogen Storage Disease Type 1 in Jordan: A Clinical Trial


 , and
, and
Abstract
1. Introduction
- Biochemical blood tests, including blood glucose levels, triglycerides, cholesterol, lactic acid and uric acid, are carried out to determine to what extent a patient sticks to the prescribed diet.
- Abdominal ultrasound to measure the volume of the liver and kidneys to assess whether or not there is an enlargement.
- Dual-energy X-ray absorptiometry (DEXA) to assess bone mineral content and vitamin D status.
- Growth parameters, including body weight and height.
2. Materials and Methods
2.1. Study Design
2.2. Patient Recruitment
2.3. Inclusion and Exclusion Criteria
- Positive family history of the condition as long as GSD has an autosomal recessive inheritance pattern.
- Biochemical findings, including hypoglycaemia, hyperlipidaemia, lactic acidosis, hyperuricemia and neutropenia, which are consistent with 1b only can be used as a tool to distinguish between the two subtypes, 1a and 1b.
- Physical considerations such as a rounded doll face, distended abdomen and short stature.
- Liver biopsy findings that show huge amounts of glycogen within the hepatocytes.
- Genetic testing that rules out the glycogen storage disease panel so that either mutated gene can be obtained: G6PC gene for 1a or SLC37A4 gene for 1b.
2.4. Questionnaire
2.5. Anthropometry and Body Composition Analysis
2.6. Dietary Intervention
- First, energy needs should be distributed throughout the day according to height, body weight and physical activity (complex carbohydrates 60–70%, proteins 15–20% and fats less than 30%).
- Second, milk formulas used must not contain sucrose, lactose or fructose.
- Third, infants who are breastfed are allowed to continue unless they are metabolically uncontrolled. Their feeding intervals must be every two to three hours, day and night, to keep blood glucose levels stable.
- Fourth, the diet should be lactose-, fructose- and sucrose-free or intake is to be severely restricted.
- Fifth, measured amounts of UCCS, according to the patient’s age and body weight. Between 1.75 and 2.5 g/kg of body weight every 4–6 h for children over two years and 1.6 g/kg of body weight every 3–4 h for young children from six months up to two years old, to be administered with water or non-acidic fluids. Acidity will break down the glycosidic bonds by which starch loses its performance at a ratio of 1:2, which gives a slow release of glucose to maintain euglycemia (≥70 mg/dL or 4 mmol/L).
- Sixth, MCT oil (0.16–0.44 g/kg and day for 32–40 months) is prescribed starting from the day of diagnosis based on the physician’s prescription.
- Finally, sucrose and lactose-free vitamin and mineral supplementations are prescribed by the metabolic physician to ensure optimal nutrition intake. (This diet is not nutritionally well-balanced regarding the amounts of certain vitamins and minerals. Therefore, patients are recommended to consume multivitamins and minerals, especially calcium and vitamin D2.).
2.7. Clinical Assessment Sheet
2.8. Blood Samples Collection
2.9. Statistical Analyses
3. Results
3.1. Socio-Demographic Characteristics of the Study Population
3.2. Laboratory Results Analysis and Abdominal Ultrasound
3.3. Nutritional Behaviours of the Study Population
3.4. Medical Information and Other GSD-Related Issues
3.5. Anthropometric Measurements and Body Composition Analysis
3.5.1. Anthropometric Measurements
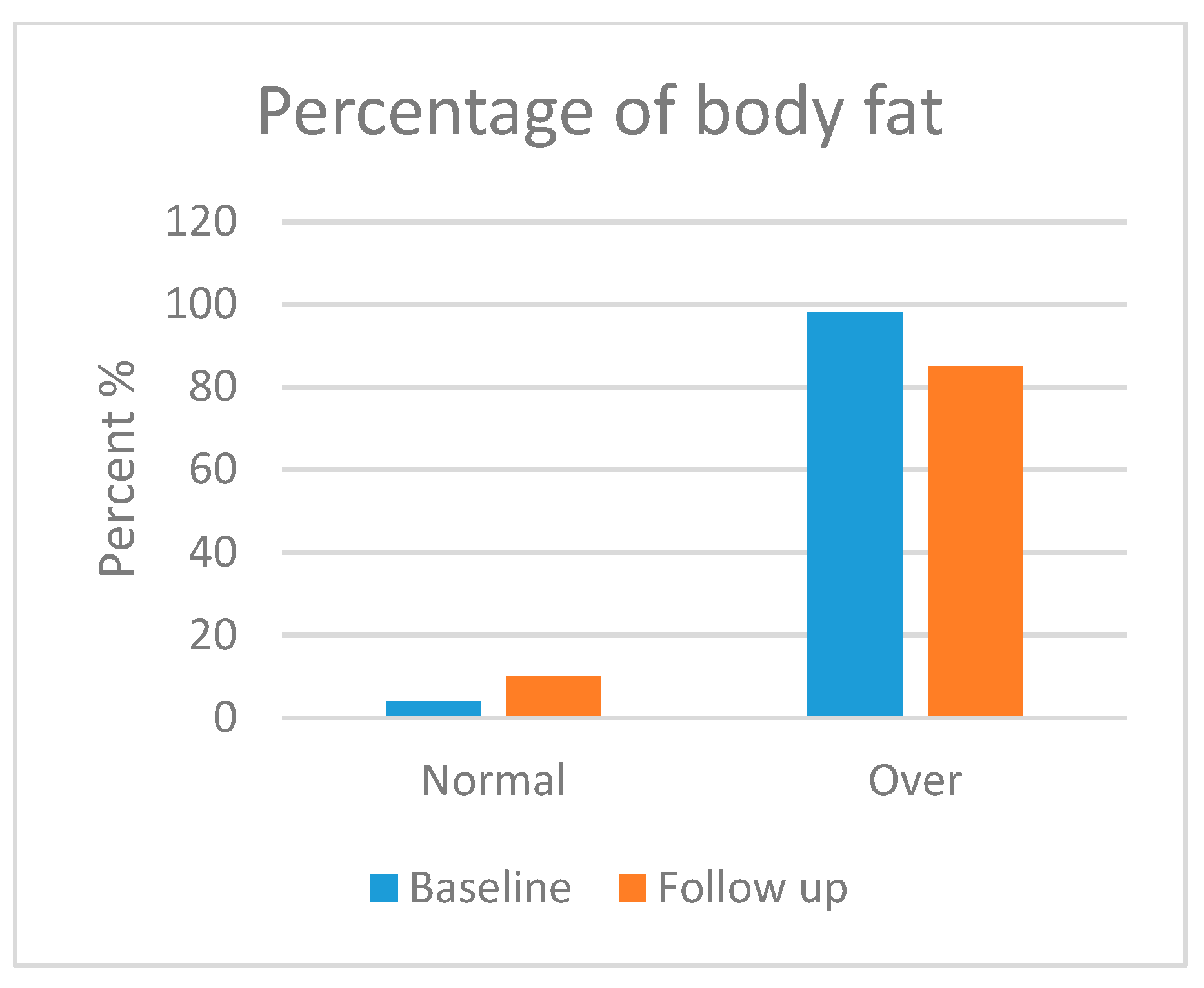
3.5.2. Body Composition Analysis
4. Discussion
- First, as a substrate for the pentose phosphate pathway (PPP), which, in turn, increases the catabolic process of ribose-5-phosphate to produce uric acid, causing hyperuricemia, which may predispose, if left untreated, to gout.
- Second, this greater flux of G6P enters the glycolytic pathway to produce huge amounts of pyruvate, leading to an alternative route that produces lactate under the action of lactate dehydrogenase (LDH), which results in lactic acidosis (hyperlactatemia).
- Third, pyruvate enters the Krebs cycle and produces acetyl CoA. Then, the latter starts lipogenesis, i.e., forming cholesterol and fatty acids. These fatty acids bind to glycerol (from glycolysis) to synthesise triglycerides, thus leading to hyperlipidaemia [42], as well as the production of malonyl CoA, which inhibits the key enzyme in β oxidation, carnitine palmitoyl transferase I (CPT I) [14,43].
5. Limitations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ellingwood, S.; Cheng, A. Biochemical and clinical aspects of glycogen storage diseases. J. Endocrinol. 2018, 238, R131–R141. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Weinstein, D. Glycogen storage diseases: Diagnosis, treatment and outcome. Transl. Sci. Rare Dis. 2016, 1, 45–72. [Google Scholar] [CrossRef]
- Derks, T.G.; Nemeth, A.; Adrian, K.; Arnell, H.; Roskjær, A.B.; Beijer, E.; Te Boekhorst, S.; Heidenborg, C.; Landgren, M.; Nilsson, M.; et al. Hepatic glycogen storage diseases: Toward one global collaborative network. J. Inborn Errors. Metab. Screen. 2017, 5, 2326409817733009. [Google Scholar] [CrossRef]
- Beyzaei, Z.; Geramizadeh, B. Molecular diagnosis of glycogen storage disease type I: A review. EXCLI J. 2019, 18, 30–46. [Google Scholar] [PubMed]
- Ross, K.M.; Ferrecchia, I.A.; Dahlberg, K.R.; Dambska, M.; Ryan, P.T.; Weinstein, D.A. Dietary Management of the glycogen storage diseases: Evolution of treatment and ongoing controversies. Adv. Nutr. 2020, 11, 439–446. [Google Scholar] [CrossRef]
- Bali, D.S.; El-Gharbawy, A.; Austin, S.; Pendyal, S.; Kishnani, P.S. Glycogen Storage Disease Type I; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Kasapkara, Ç.S.; Demir, G.C.; Hasanoğlu, A.; Tümer, L. Continuous glucose monitoring in children with glycogen storage disease type I. Eur. J. Clin. Nutr. 2014, 68, 101–105. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Austin, S.L.; Abdenur, J.E.; Arn, P.; Bali, D.S.; Boney, A.; Chung, W.K.; Dagli, A.I.; Dale, D.; Koeberl, D.; et al. Diagnosis and management of glycogen storage disease type I: A practice guideline of the American College of Medical Genetics and Genomics. Genet. Med. 2014, 16, e1–e29. [Google Scholar] [CrossRef] [PubMed]
- NORD. Glycogen Storage Disease Type I. 2019. Available online: https://rarediseases.org/rare-diseases/glycogen-storage-disease-type-i/ (accessed on 2 January 2020).
- Raza, M.; Arif, F.; Giyanwani, P.R.; Azizullah, S.; Kumari, S. Dietary therapy for Von Gierke’s disease: A Case Report. Cureus 2017, 9, e1548. [Google Scholar] [CrossRef]
- Sim, S.W.; Weinstein, D.A.; Lee, Y.M.; Jun, H.S. Glycogen storage disease type Ib: Role of glucose-6-phosphate transporter in cell metabolism and function. FEBS Lett. 2020, 594, 3–18. [Google Scholar] [CrossRef]
- Kanungo, S.; Wells, K.; Tribett, T.; El-Gharbawy, A. Glycogen metabolism and glycogen storage disorders. Ann. Transl. Med. 2018, 6, 474. [Google Scholar] [CrossRef]
- Burda, P.; Hochuli, M. Hepatic glycogen storage disorders: What have we learned in recent years? Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Derks, T.G.; van Rijn, M. Lipids in hepatic glycogen storage diseases: Pathophysiology, monitoring of dietary management and future directions. J. Inherit. Metab. Dis. 2015, 38, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, D.A.; Steuerwald, U.; De Souza, C.F.; Derks, T.G.J. Inborn errors of metabolism with hypoglycemia: Glycogen storage diseases and inherited disorders of gluconeogenesis. Pediatr. Clin. N. Am. 2018, 65, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.M.; Silva, N.J.; Dias, P.G.; Porto, J.F.C.; Santos, L.C.; Costa, J.M.N. Glycogen storage disease type 1a–a secondary cause for hyperlipidemia: Report of five cases. J. Diabetes Metab. Disord. 2013, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.Y.; Kim, G.Y.; Cho, J.H. Recent development and gene therapy for glycogen storage disease type Ia. Liver Res. 2017, 1, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Das, A.M.; Lücke, T.; Meyer, U.; Hartmann, H.; Illsinger, S. Glycogen storage disease type 1: Impact of medium-chain triglycerides on metabolic control and growth. Ann. Nutr. Metab. 2010, 56, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Arshad, H.; Alvi, A.; Suleman, H. Clinical presentation and biochemical findings in children with glycogen storage disease type 1a. Pak. Armed Forces Med. J. 2015, 65, 682–685. [Google Scholar]
- Lee, Y.M.; Conlon, T.J.; Specht, A.; Coleman, K.E.; Brown, L.M.; Estrella, A.M.; Dambska, M.; Dahlberg, K.R.; Weinstein, D.A. Long-term safety and efficacy of AAV gene therapy in the canine model of glycogen storage disease type Ia. J. Inherit. Metab. Dis. 2018, 41, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.; Wartchow, E.; Mierau, G. Glycogen storage diseases: A brief review and update on clinical features, genetic abnormalities, pathologic features, and treatment. Ultrastruct. Pathol. 2011, 35, 183–196. [Google Scholar] [CrossRef]
- Sundaram, S.S.; Alonso, E.M. Abnormalities of carbohydrate metabolism and the liver. In Pediatric Gastrointestinal and Liver Disease; Elsevier Inc.: Amsterdam, The Netherlands, 2006; pp. 913–924. [Google Scholar]
- Rossi, A.; Ruoppolo, M.; Formisano, P.; Villani, G.; Albano, L.; Gallo, G.; Crisci, D.; Moccia, A.; Parenti, G.; Strisciuglio, P.; et al. Insulin-resistance in glycogen storage disease type Ia: Linking carbohydrates and mitochondria? J. Inherit. Metab. Dis. 2018, 41, 985–995. [Google Scholar] [CrossRef]
- Minarich, L.A.; Kirpich, A.; Fiske, L.M.; Weinstein, D.A. Bone mineral density in glycogen storage disease type Ia and Ib. Genet. Med. 2012, 14, 737–741. [Google Scholar] [CrossRef]
- Beegle, R.D.; Brown, L.M.; Weinstein, D.A. Regression of hepatocellular adenomas with strict dietary therapy in patients with glycogen storage disease type I. In JIMD Reports; Zschocke, J., Baumgartner, M., Morava, E., Patterson, M., Rahman, S., Peters, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 18, pp. 23–32. [Google Scholar] [CrossRef]
- Dambska, M.; Labrador, E.B.; Kuo, C.L.; Weinstein, D.A. Prevention of complications in glycogen storage disease type Ia with optimization of metabolic control. Pediatr. Diabetes 2017, 18, 327–331. [Google Scholar] [CrossRef]
- Melis, D.; Rossi, A.; Pivonello, R.; Salerno, M.; Balivo, F.; Spadarella, S.; Muscogiuri, G.; Casa, R.D.; Formisano, P.; Andria, G.; et al. Glycogen storage disease type Ia (GSDIa) but not glycogen storage disease type Ib (GSDIb) is associated to an increased risk of metabolic syndrome: Possible role of microsomal glucose 6-phosphate accumulation. Orphanet. J. Rare Dis. 2015, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Assiri, Y.M.; Iqbal, M.M.; Almanie, R.A.; Alotaibi, A.; Alharbi, F.; Al Jobran, B.; Abbag, H.; Almoghrabi, M.; Jarad, F.; Asiri, A.; et al. Glycogen storage disease in pediatric population. Egypt. J. Hosp. Med. 2018, 70, 1539–1543. [Google Scholar] [CrossRef]
- Rake, J.; Visser, G.; Labrune, P.; Leonard, J.; Ullrich, K.; Smit, P. Glycogen storage disease type I: Diagnosis, management, clinical course and outcome. Results of the European study on glycogen storage disease type I (ESGSD I). Eur. J. Pediatr. 2002, 161, S20–S34. [Google Scholar] [CrossRef] [PubMed]
- Parikh, N.S.; Ahlawat, R. Glycogen Storage Disease Type I. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK534196/ (accessed on 1 February 2022).
- Rake, J.; Visser, G.; Labrune, P.; Leonard, J.; Ullrich, K.; Smit, P. Guidelines for management of glycogen storage disease type I–European study on glycogen storage disease type I (ESGSD I). Eur. J. Pediatr. 2002, 161, S112–S119. [Google Scholar] [CrossRef]
- Boyer, S.W.; Barclay, L.J.; Burrage, L.C. Inherited metabolic disorders: Aspects of chronic nutrition management. Nutr. Clin. Pract. 2015, 30, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Derks, T.G.; Martens, D.H.; Sentner, C.P.; van Rijn, M.; de Boer, F.; Smit, G.P.; van Spronsen, F.J. Dietary treatment of glycogen storage disease type Ia: Uncooked cornstarch and/or continuous nocturnal gastric drip-feeding? Mol. Genet. Metab. 2013, 109, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Froissart, R.; Piraud, M.; Boudjemline, A.M.; Vianey-Saban, C.; Petit, F.; Hubert-Buron, A.; Eberschweiler, P.T.; Gajdos, V.; Labrune, P. Glucose-6-phosphatase deficiency. Orphanet. J. Rare Dis. 2011, 6, 27. [Google Scholar] [CrossRef]
- Jeong, Y.J.; Kang, B.; Choi, S.Y.; Ki, C.S.; Lee, S.Y.; Park, H.D.; Choe, Y.H. Does type I truly dominate hepatic glycogen storage diseases in Korea?: A single center study. Pediatr. Gastroenterol. Hepatol. Nutr. 2014, 17, 239–247. [Google Scholar] [CrossRef][Green Version]
- Santos, B.L.; de Souza, C.F.; Schuler-Faccini, L.; Refosco, L.; Epifanio, M.; Nalin, T.; Vieira, S.M.G.; Schwartz, I.V.D. Glycogen storage disease type I: Clinical and laboratory profile. J. Pediatr. 2014, 90, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Steunenberg, T.A.; Peeks, F.; Hoogeveen, I.J.; Mitchell, J.J.; Mundy, H.; de Boer, F.; Lubout, C.M.A.; de Souza, C.F.; Weinstein, D.A.; Derks, T.G.J. Safety issues associated with dietary management in patients with hepatic glycogen storage disease. Mol. Genet. Metab. 2018, 125, 79–85. [Google Scholar] [CrossRef] [PubMed]
- van Calcar, S. Nutrition management of glycogen storage disease type 1. In Nutrition Management of Inherited Metabolic Diseases; Bernstein, L.E., Rohr, F., Helm, J.R., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 307–317. [Google Scholar]
- Szymanska, E.; vel Emczynska-Seliga, E.E.; Rokicki, D.; Książyk, J. Body composition measurements using bioelectrical impedance analysis (BIA) in pediatric patients with hepatic glycogen storage disease–Preliminary data. Clin. Nutr. ESPEN 2017, 19, 35–37. [Google Scholar] [CrossRef]
- Shaw, V. (Ed.) Clinical Paediatric Dietetics, 4th ed.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2015; p. 554. [Google Scholar]
- Grummer-Strawn, L.; Krebs, N.F.; Reinold, C.M. Use of World Health Organization and CDC Growth Charts for Children Aged 0-59 Months in the United States. MMWR Surveill Summ. 2010, 59, 1–15. Available online: https://stacks.cdc.gov/view/cdc/5746 (accessed on 2 May 2023).
- The Study Spot. Von Gierke (Glycogen Storage Disease 1) [Video File]. 14 December 2014. Available online: https://youtu.be/p4dU4deqssw (accessed on 2 January 2023).
- Bandsma, R.H.; Smit, P.G.; Kuipers, F. Disturbed lipid metabolism in glycogen storage disease type 1. Eur. J. Pediatr. 2002, 161, S65–S69. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.Y.; Cho, J.H.; Kim, G.Y.; Mansfield, B.C. Molecular biology and gene therapy for glycogen storage disease type Ib. J. Inherit. Metab. Dis. 2018, 41, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.S.; Weinstein, D.A.; Lee, Y.M.; Mansfield, B.C.; Chou, J.Y. Molecular mechanisms of neutrophil dysfunction in glycogen storage disease type Ib. Blood 2014, 123, 2843–2853. [Google Scholar] [CrossRef]
- Li, R.R.; Mao, J.F.; Yu, K.; Wang, L. Dietary or enteral medium-chain triglyceride usage in a Chinese general hospital. Asia Pac. J. Clin. Nutr. 2015, 24, 387–393. [Google Scholar] [CrossRef]
- Shah, N.D.; Limketkai, B.N. The use of medium-chain triglycerides in gastrointestinal disorders. Pract. Gastroenterol. 2017, 160, 20–25. [Google Scholar]
- Nagasaka, H.; Hirano, K.I.; Ohtake, A.; Miida, T.; Takatani, T.; Murayama, K.; Yorifuji, T.; Kobayashi, K.; Kanazawa, M.; Oga-wa, A.; et al. Improvements of hypertriglyceridemia and hyperlacticemia in Japanese children with glycogen storage disease type Ia by medium-chain triglyceride milk. Eur. J. Pediatr. 2007, 166, 1009–1016. [Google Scholar] [CrossRef]
- Melis, D.; Minopoli, G.; Balivo, F.; Marcolongo, P.; Parini, R.; Paci, S.; Dionisi-Vici, C.; Della Casa, R.; Benedetti, A.; Andria, G.; et al. Vitamin E improves clinical outcome of patients affected by glycogen storage disease type Ib. JIMD Rep. 2015, 25, 39–45. [Google Scholar] [CrossRef]
- Tanchoco, C.C.; Cruz, A.J.; Rogaccion, J.M.; Casem, R.S.; Rodriguez, M.P.; Orense, C.L.; Hermosura, L.C. Diet supplemented with MCT oil in the management of childhood diarrhea. Asia Pac. J. Clin. Nutr. 2007, 16, 286–292. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | N | % |
|---|---|---|
| Multivitamins intake | ||
| Yes | 34 | 89.5 |
| No | 4 | 10.5 |
| Vitamin D deficiency | ||
| Yes | 23 | 60.5 |
| No | 15 | 39.5 |
| Frequency and dose of vitamin D intake | ||
| None | 2 | 5.3 |
| Daily (2000 IU *) | 10 | 26.3 |
| Every other day (5000 IU) | 6 | 15.8 |
| Weekly (50,000 IU) | 20 | 52.6 |
| Calcium supplement intake | ||
| Yes | 22 | 57.9 |
| No | 16 | 42.1 |
| GSD subtype | ||
| Type 1a | 11 | 28.9 |
| Type 1b | 27 | 71.1 |
| Initial clinical symptoms | ||
| Hypoglycaemia | 26 | 68.4 |
| Hepatomegaly | 9 | 23.7 |
| Both | 3 | 7.9 |
| Method of diagnosis | ||
| Clinical presentation | 15 | 39.5 |
| Liver biopsy and clinical presentation | 9 | 23.7 |
| Clinical presentation and positive family history | 7 | 18.4 |
| Genetic testing and positive family history | 4 | 10.5 |
| Genetic testing and clinical presentation | 3 | 7.9 |
| MCT Intervention | Mean | Normal Range | SD | Minimum | Maximum | Median | p-Value | |
|---|---|---|---|---|---|---|---|---|
| Glucose (mg/dL) | Before After | 53.03 90.47 | 70–110 | 16.619 18.569 | 13 58 | 93 142 | 55.50 88.00 | ≤0.001 b |
| TG (mg/dL) | Before After | 657.16 368.35 | 50–200 | 660.812 255.958 | 202.6 126.9 | 2895.0 1136.8 | 351.85 284.60 | ≤0.001 a |
| Cholesterol (mg/dL) | Before After | 251.16 170.21 | 70–200 | 73.486 65.876 | 122 103 | 405 383 | 249.50 143 | ≤0.001 a |
| Uric Acid (mg/dL) | Before After | 7.70 5.94 | 2.4–7.0 | 1.917 1.660 | 5.56 2.7 | 13.3 12.70 | 7.10 5.85 | ≤0.001 a |
| Lactate (mg/dL) | Before After | 53.17 19.51 | 0–25.2 | 27.030 6.533 | 13.9 10.11 | 129.00 40.40 | 41.00 18.05 | ≤0.001 a |
| Neutrophils Count (103/µL) | Before After | 1.93 1.82 | 1.5–7.0 | 1.802 1.145 | 0.10 0.22 | 6.19 4.42 | 1.06 1.5 | 0.331 a |
| HCT (%) | Before After | 31.73 32.62 | 31–54 | 4.580 4.748 | 22.1 17.2 | 42.1 47.6 | 31.25 32.15 | 0.020 a |
| Laboratories Tests | Abnormal Levels before MCT Oil (%) | Abnormal Levels after MCT Oil (%) | p-Value |
|---|---|---|---|
| Glucose (mg/dL) | 94 | 7.9 | 0.001 |
| Lactic Acid (mg/dL) | 94.7 | 18.4 | 0.001 |
| Triglycerides (mg/dL) | 100 | 71.1 | 0.001 |
| Cholesterol (mg/dL) | 73.7 | 21.1 | 0.001 |
| Uric Acid (mg/dL) | 97.4 | 52.6 | 0.001 |
| Neutrophils (103/µL) | 60.5 | 55.3 | 0.001 |
| Variable | Baseline | Follow-Up | ||||
|---|---|---|---|---|---|---|
| Age Group | Mean | SD | Mean | SD | p-Value | |
| Weight-for-age | <2 years ≥2 years | 36.67 25.68 | 41.826 29.014 | 39.17 26.14 | 48.008 30.101 | 0.593 a 0.940 a |
| Height-for-age | <2 years ≥2 years | 33.27 4.13 | 46.708 8.351 | 33.13 5.25 | 36.258 10.328 | 1.00 a 0.034 a |
| BMI-for-age | <2 years ≥2 years | 45.53 75.34 | 27.632 20.487 | 62.90 71.63 | 42.062 23.503 | 0.593 a 0.074 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subih, H.S.; Qudah, R.A.; Janakat, S.; Rimawi, H.; Elsahoryi, N.A.; Alyahya, L. Medium-Chain Triglyceride Oil and Dietary Intervention Improved Body Composition and Metabolic Parameters in Children with Glycogen Storage Disease Type 1 in Jordan: A Clinical Trial. Foods 2024, 13, 1091. https://doi.org/10.3390/foods13071091
Subih HS, Qudah RA, Janakat S, Rimawi H, Elsahoryi NA, Alyahya L. Medium-Chain Triglyceride Oil and Dietary Intervention Improved Body Composition and Metabolic Parameters in Children with Glycogen Storage Disease Type 1 in Jordan: A Clinical Trial. Foods. 2024; 13(7):1091. https://doi.org/10.3390/foods13071091
Chicago/Turabian StyleSubih, Hadil S., Reem A. Qudah, Sana Janakat, Hanadi Rimawi, Nour Amin Elsahoryi, and Linda Alyahya. 2024. "Medium-Chain Triglyceride Oil and Dietary Intervention Improved Body Composition and Metabolic Parameters in Children with Glycogen Storage Disease Type 1 in Jordan: A Clinical Trial" Foods 13, no. 7: 1091. https://doi.org/10.3390/foods13071091
APA StyleSubih, H. S., Qudah, R. A., Janakat, S., Rimawi, H., Elsahoryi, N. A., & Alyahya, L. (2024). Medium-Chain Triglyceride Oil and Dietary Intervention Improved Body Composition and Metabolic Parameters in Children with Glycogen Storage Disease Type 1 in Jordan: A Clinical Trial. Foods, 13(7), 1091. https://doi.org/10.3390/foods13071091