
Analysis of the Changes in Physicochemical Properties and Microbial Communities During Fermentation of Sweet Fermented Rice


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Instruments and Equipment
2.3. Methods
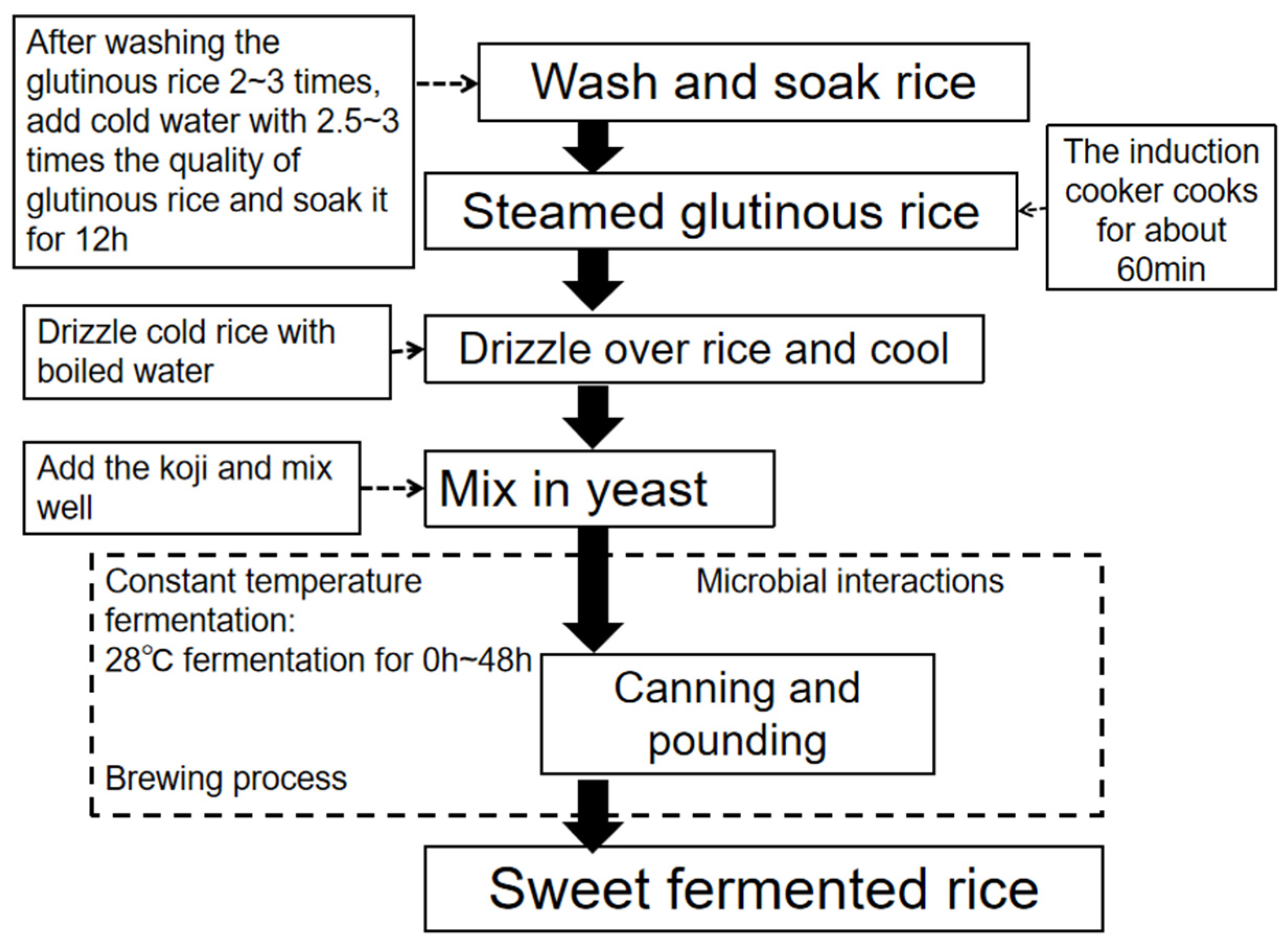
2.3.1. Sample Preparation and Collection
2.3.2. Evaluation of Flavor and Determination of Physicochemical Properties
2.3.3. High-Throughput Sequencing and Sequence Processing Analysis
2.3.4. Species Composition Assessment
2.3.5. Alpha Diversity and Beta Diversity Assessment
2.3.6. Analysis at the Functional Level
3. Results
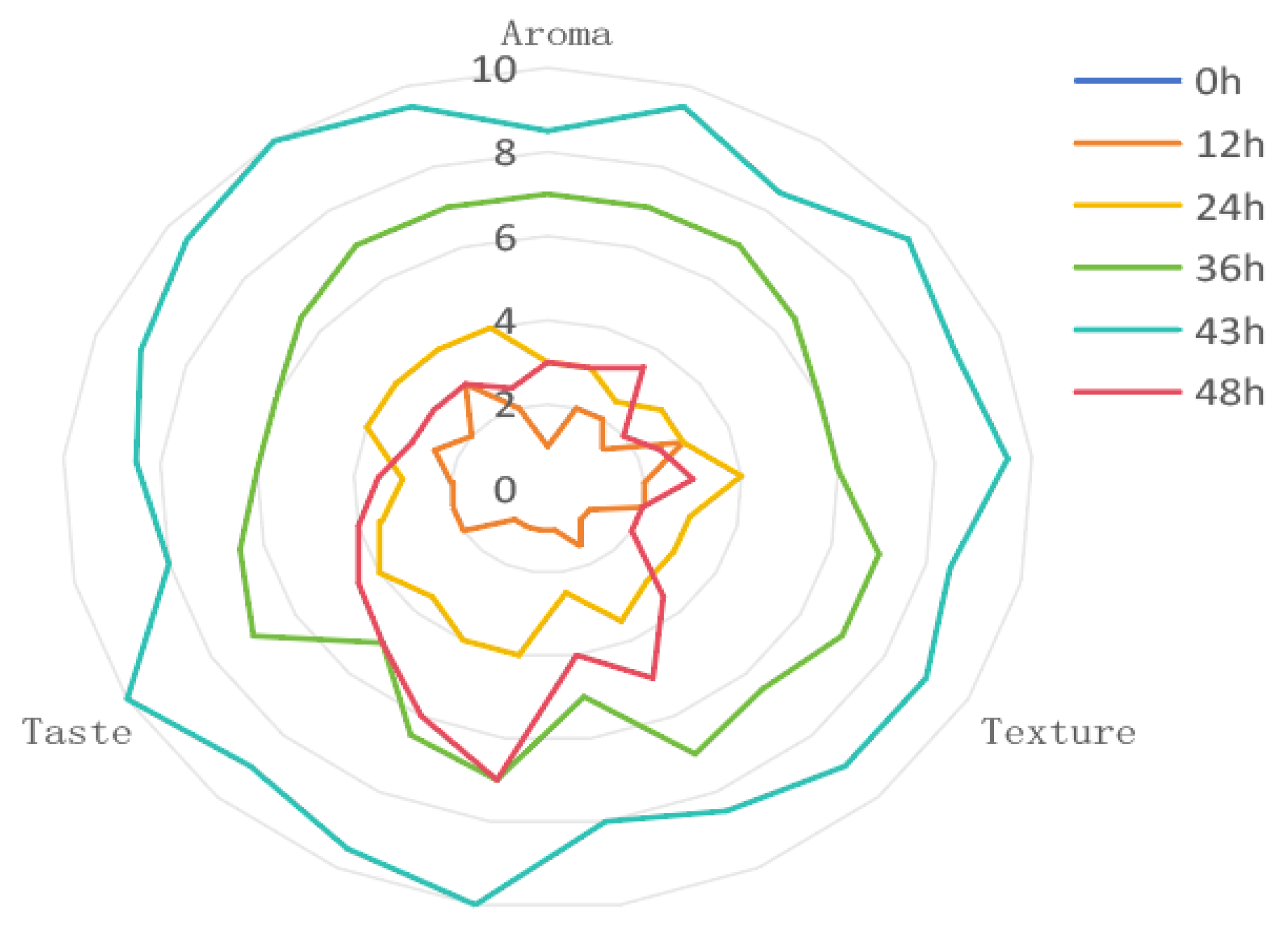
3.1. Evaluation of Flavor and Analysis of Physicochemical Properties
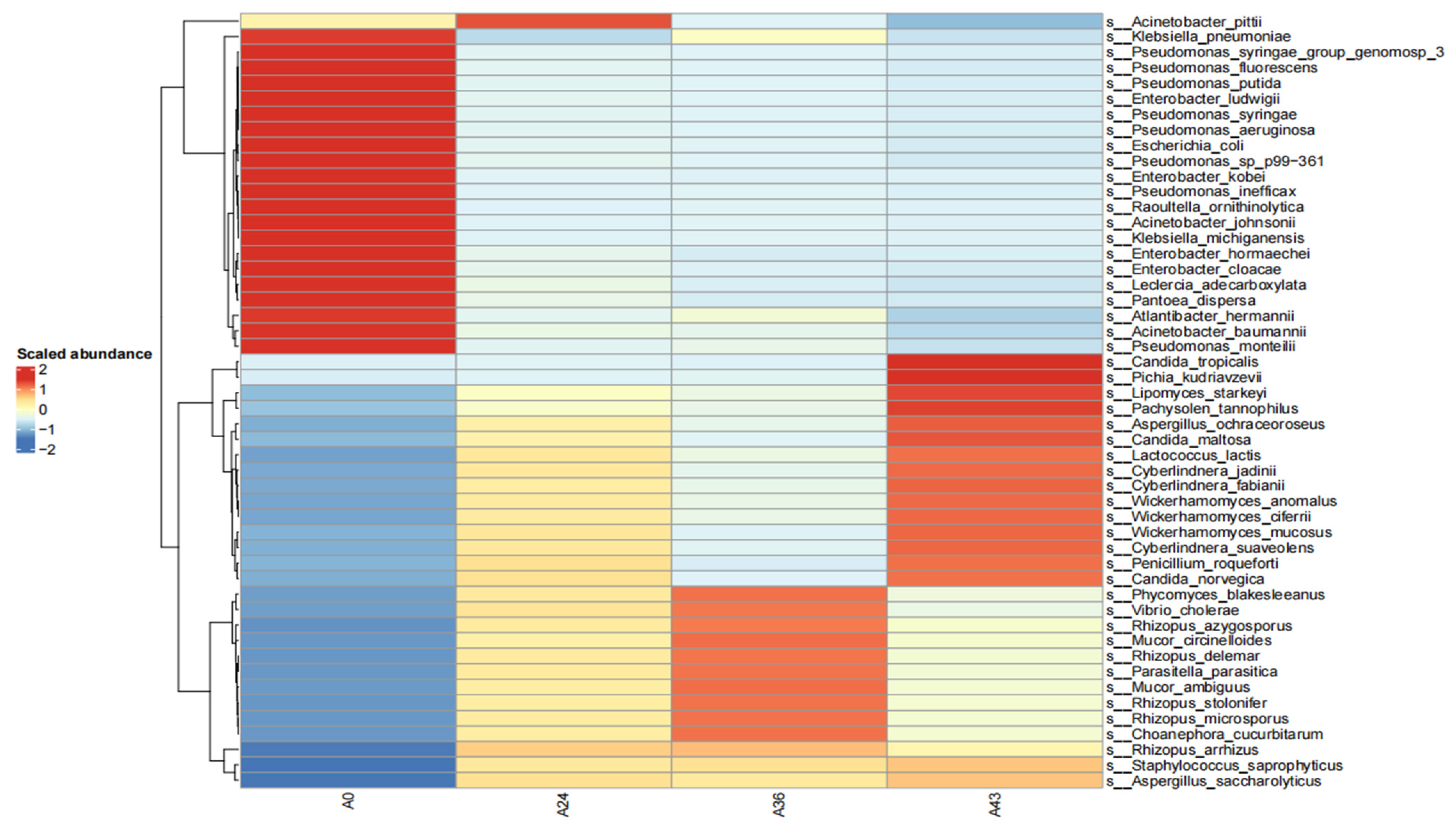
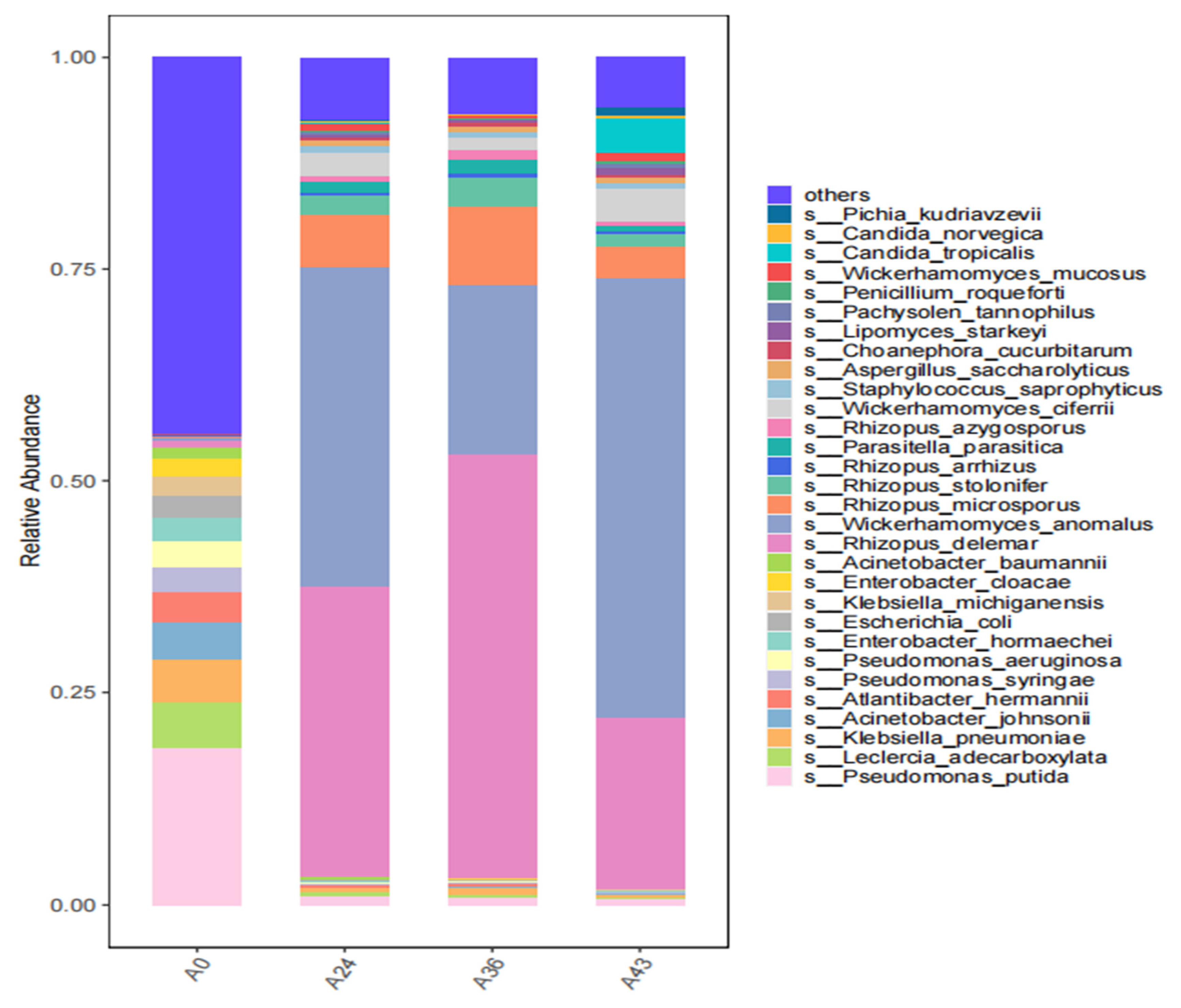
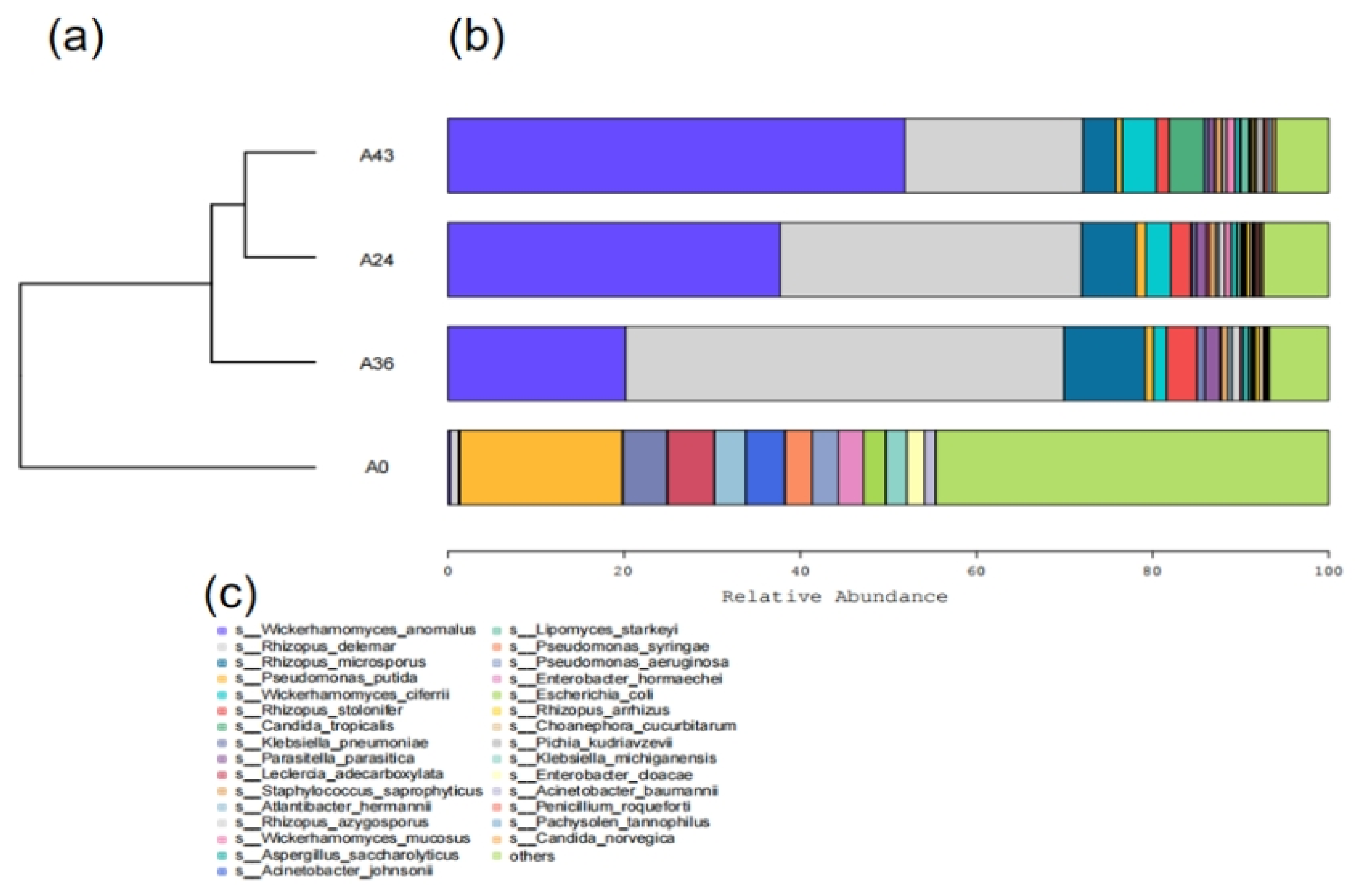
3.2. Species Composition Analysis
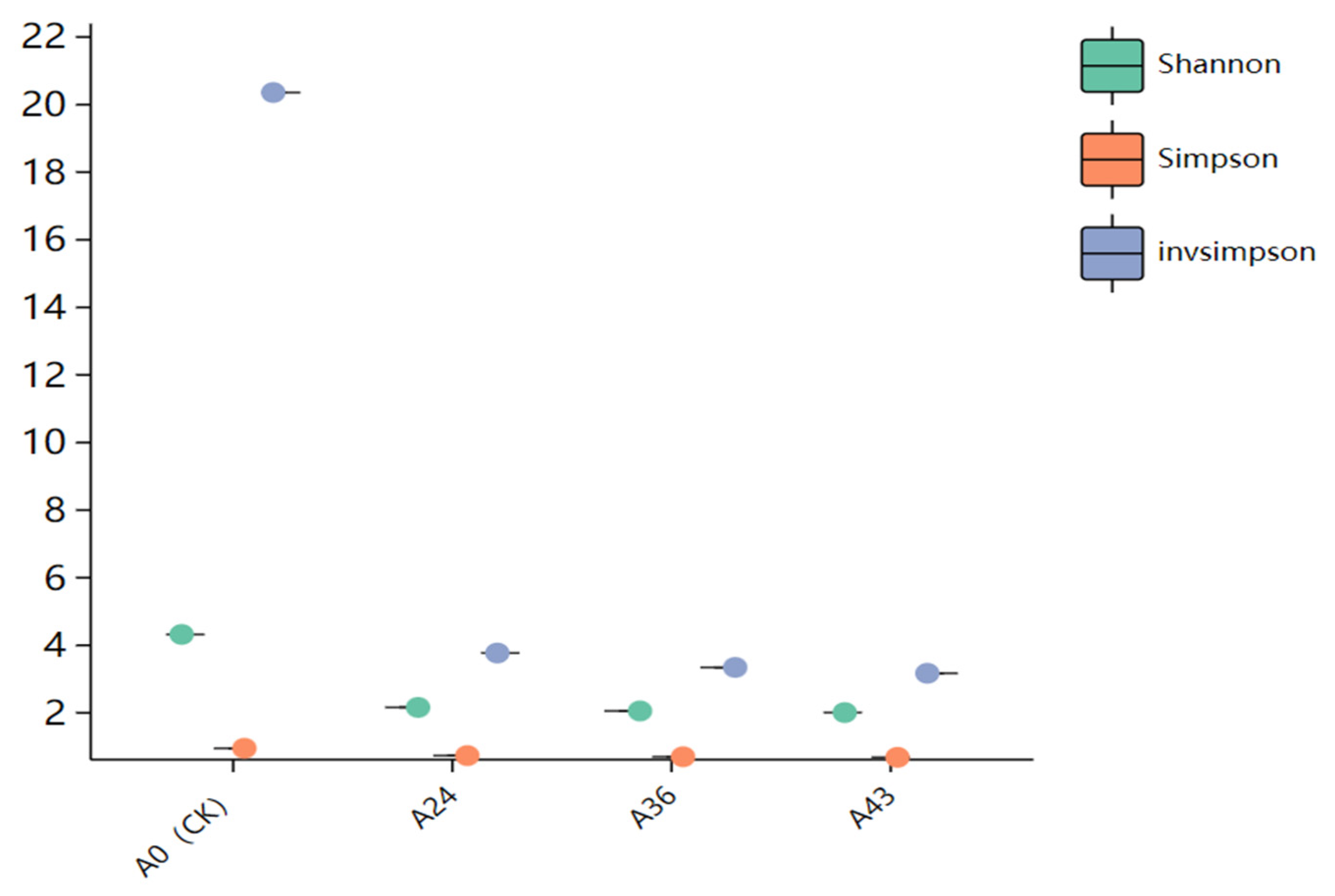
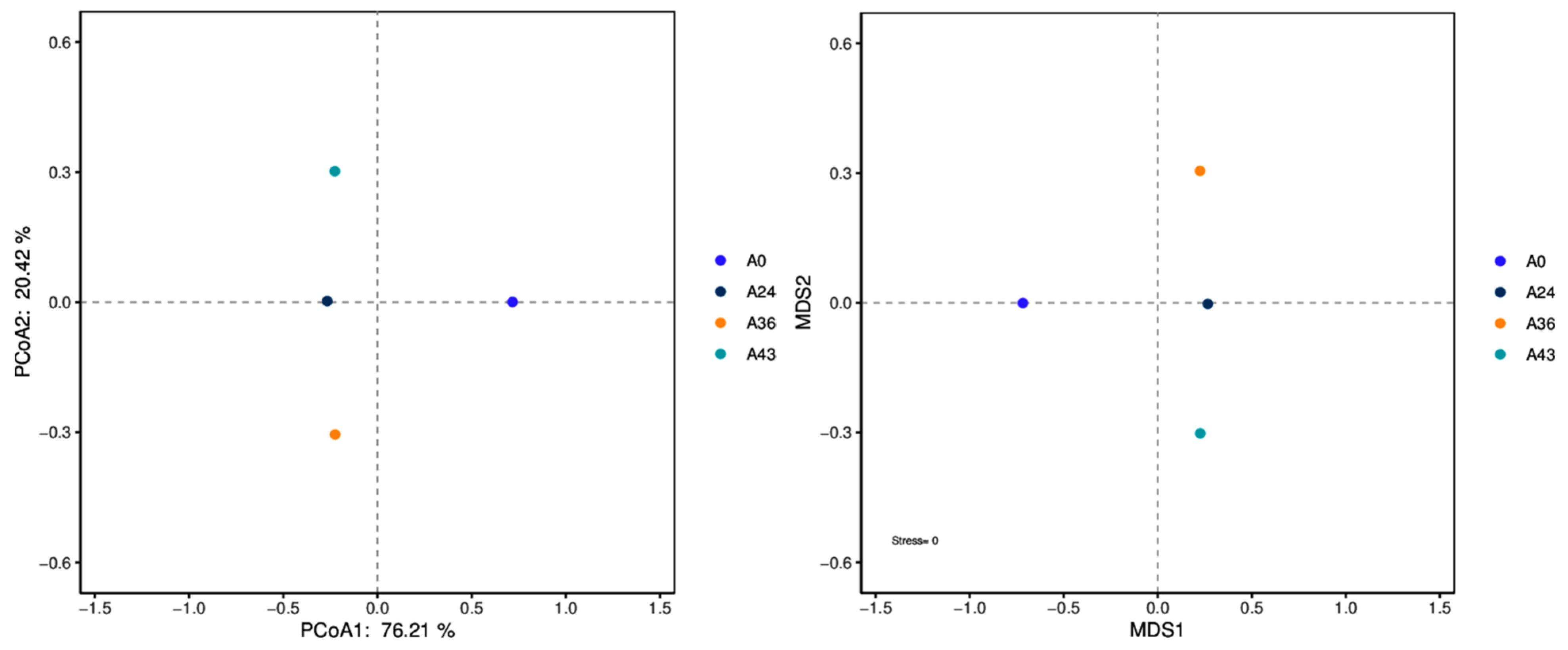
3.3. Alpha Diversity and Beta Diversity Analysis
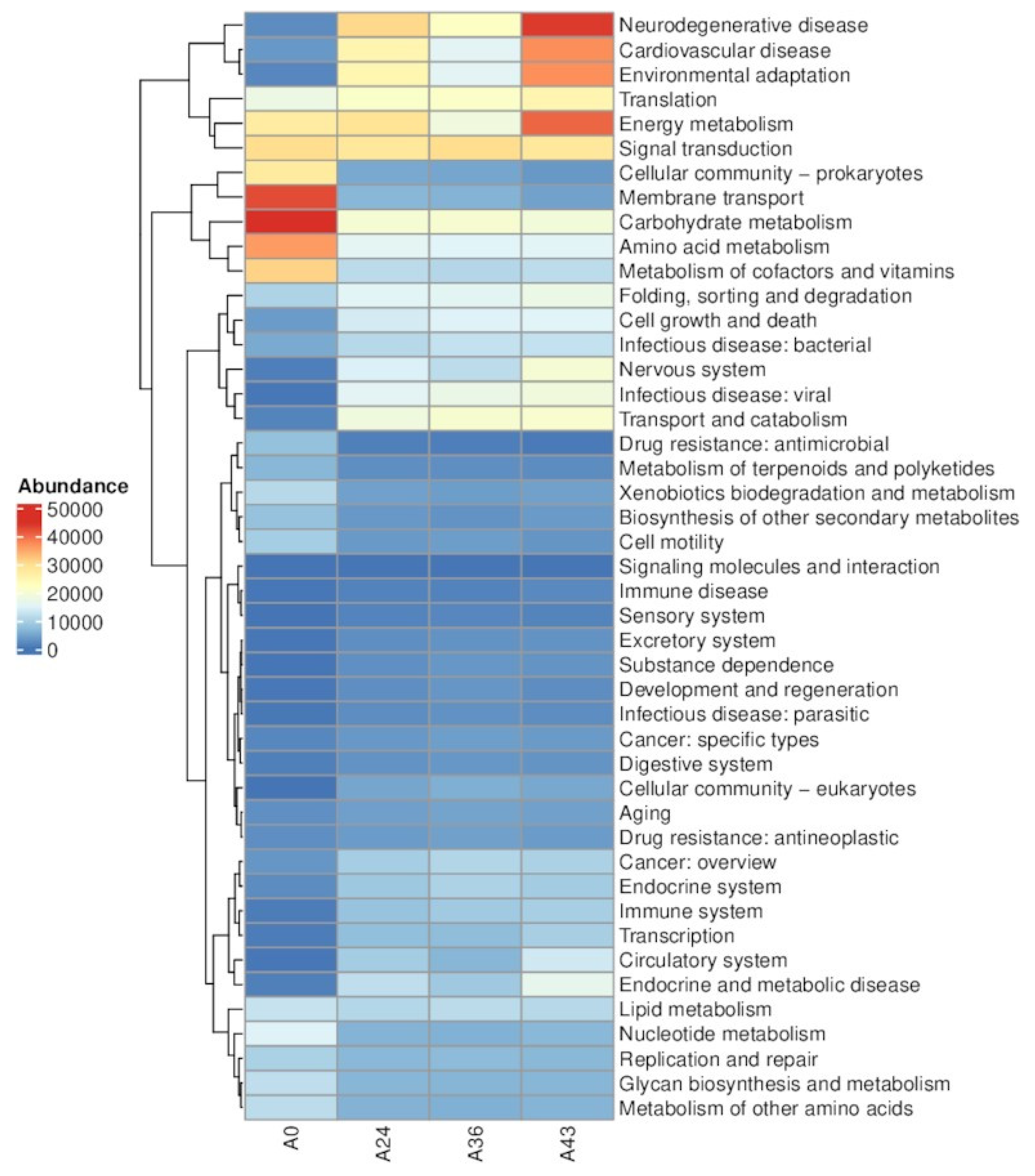
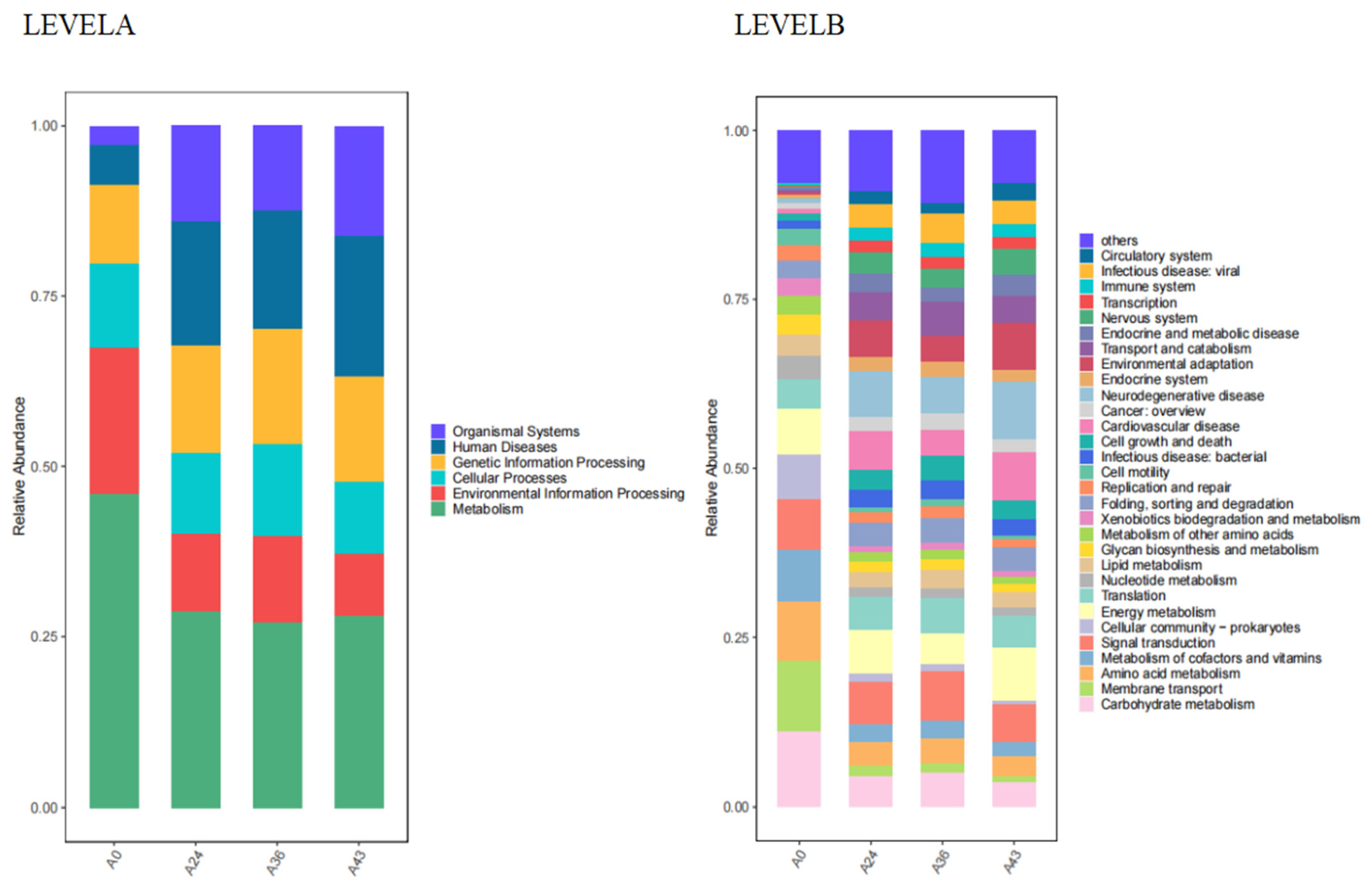
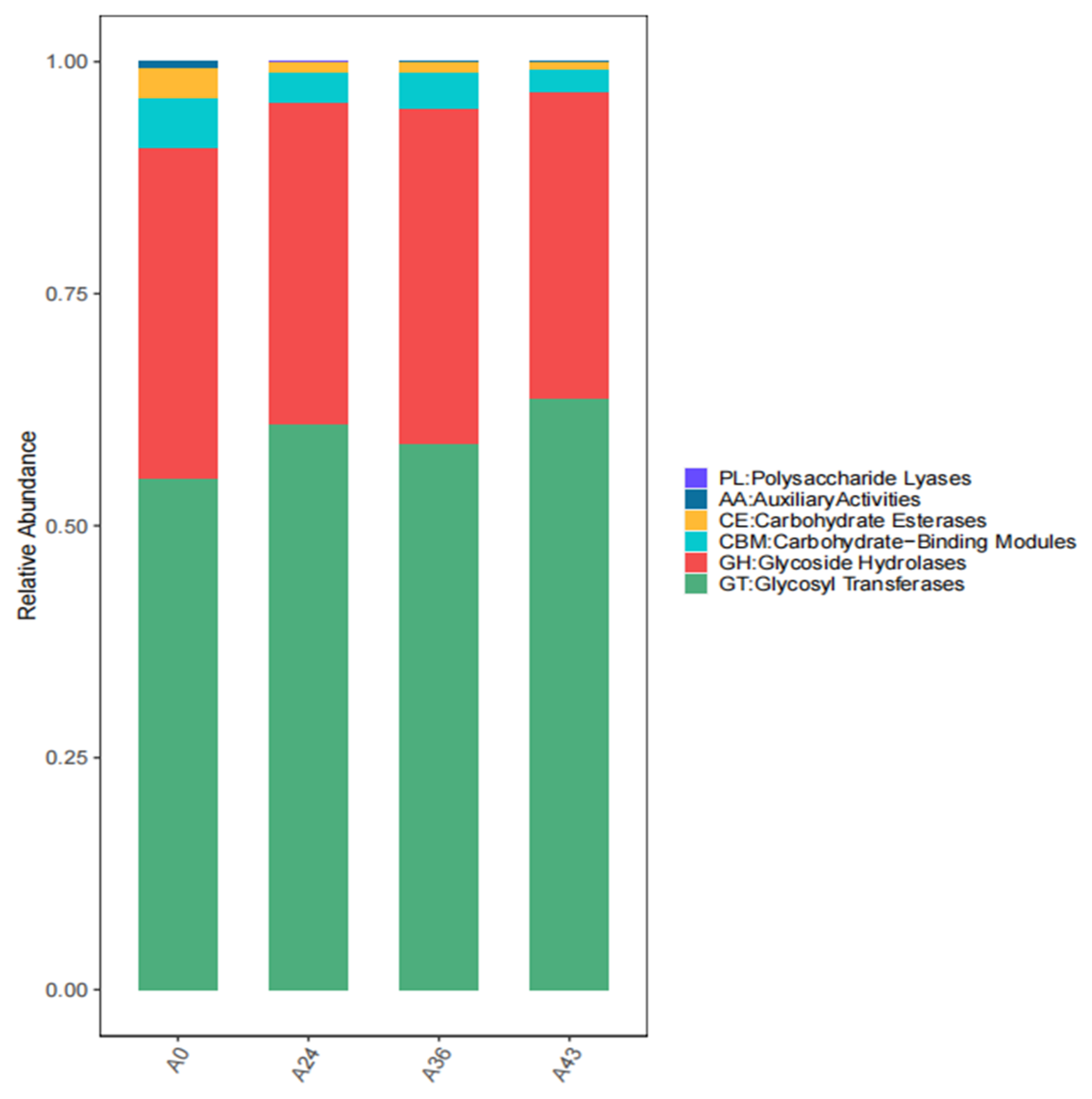
3.4. Functional Level Analysis
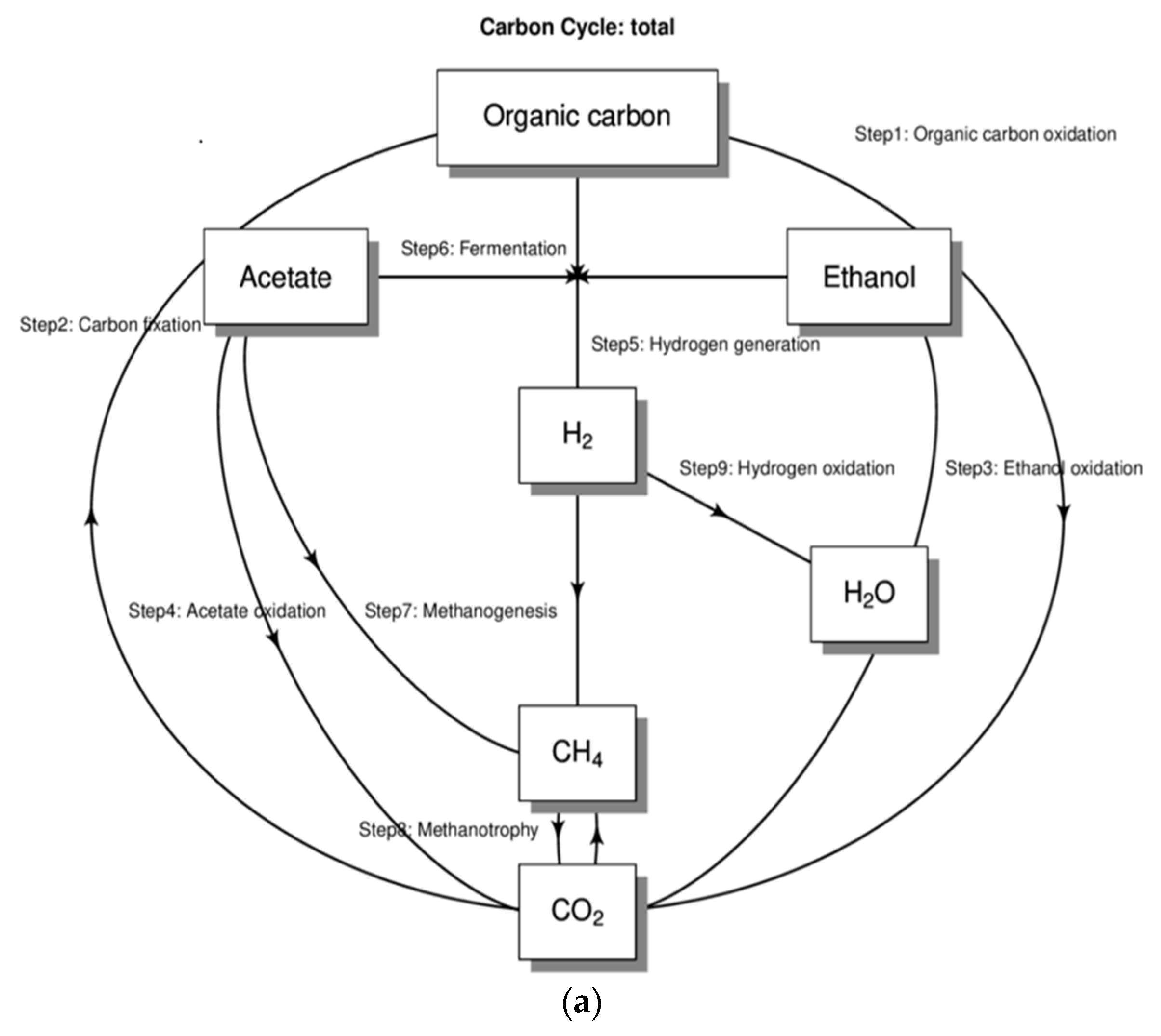
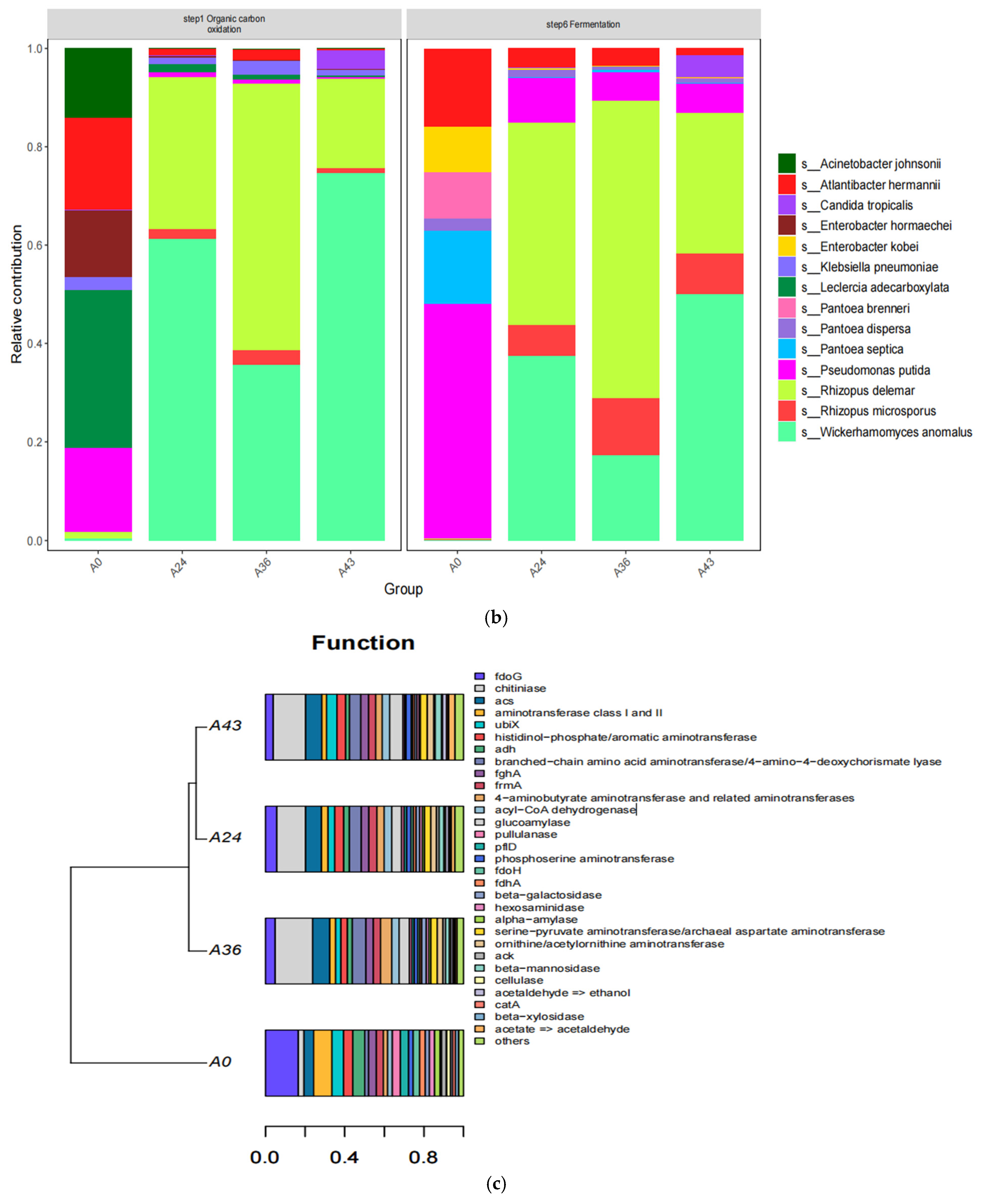
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LEVELB | A0 | A24 | A36 | A43 |
|---|---|---|---|---|
| Aging | 2675.92 | 4147.91 | 4599.61 | 4305.97 |
| Amino acid metabolism | 36,127.18 | 15,742.45 | 15,160.08 | 15,219.15 |
| Biosynthesis of other secondary metabolites | 7918.37 | 3532.37 | 3006.32 | 3752.05 |
| Cancer: overview | 3266.71 | 9320.59 | 10626.61 | 10,000.54 |
| Cancer: specific types | 1757.93 | 3525.66 | 4051.72 | 3722.13 |
| Carbohydrate metabolism | 44,808.81 | 19,870.03 | 20,310.00 | 18,963.77 |
| Cardiovascular disease | 3519.72 | 24,856.46 | 15,587.31 | 37,271.02 |
| Cell growth and death | 3845.75 | 13,802.35 | 14,767.09 | 15,110.44 |
| Cell motility | 9327.21 | 3659.88 | 3987.70 | 3195.53 |
| Cellular community - eukaryotes | 32.51 | 4845.36 | 5791.89 | 5079.86 |
| Cellular community - prokaryotes | 27,492.73 | 5211.41 | 4854.55 | 3495.79 |
| Circulatory system | 233.32 | 9238.22 | 6541.71 | 13,317.83 |
| Development and regeneration | 378.22 | 2555.40 | 3315.84 | 2419.89 |
| Digestive system | 1117.77 | 2947.24 | 3439.31 | 3049.20 |
| Drug resistance: antimicrobial | 7731.89 | 1070.23 | 1025.40 | 586.61 |
| Drug resistance: antineoplastic | 2577.31 | 3777.11 | 4376.20 | 3773.51 |
| Endocrine and metabolic disease | 1089.50 | 11,758.62 | 8854.67 | 16,572.83 |
| Endocrine system | 2225.45 | 8709.26 | 10,016.93 | 9138.44 |
| Energy metabolism | 27,196.95 | 28,906.73 | 18,505.78 | 40,458.00 |
| Environmental adaptation | 1848.96 | 24,725.68 | 15,581.02 | 37,154.49 |
| Excretory system | 182.32 | 2618.73 | 3028.19 | 2976.12 |
| Folding, sorting and degradation | 10,113.97 | 15,306.08 | 15,680.13 | 17,494.11 |
| Glycan biosynthesis and metabolism | 11,811.23 | 6527.91 | 6448.22 | 6550.90 |
| Immune disease | 276.05 | 1341.12 | 1322.05 | 1965.53 |
| Immune system | 807.53 | 8053.24 | 8946.12 | 9502.07 |
| Infectious disease: bacterial | 5329.26 | 10,977.55 | 12,172.77 | 12,166.27 |
| Infectious disease: parasitic | 330.19 | 2392.15 | 2931.09 | 2349.06 |
| Infectious disease: viral | 258.51 | 15,515.05 | 17,403.48 | 18,757.58 |
| Lipid metabolism | 12,328.97 | 10,651.60 | 11,422.98 | 10,977.24 |
| Membrane transport | 42,630.88 | 6636.62 | 6189.63 | 4395.03 |
| Metabolism of cofactors and vitamins | 31,123.23 | 11,405.85 | 10,693.61 | 11,623.45 |
| Metabolism of other amino acids | 11,489.73 | 6124.54 | 5911.47 | 6325.28 |
| Metabolism of terpenoids and polyketides | 6607.23 | 2529.87 | 2571.41 | 2266.63 |
| Nervous system | 1007.23 | 14,527.07 | 11,521.70 | 19,930.87 |
| Neurodegenerative disease | 2299.48 | 30,618.01 | 21,817.52 | 44,002.95 |
| Nucleotide metabolism | 14,738.38 | 6269.23 | 5988.87 | 6694.13 |
| Replication and repair | 9893.67 | 6684.42 | 6981.63 | 6661.09 |
| Sensory system | 6.81 | 1482.42 | 1821.13 | 1466.52 |
| Signal transduction | 30,004.02 | 28,074.00 | 29,988.59 | 28,270.69 |
| Signaling molecules and interaction | 0.64 | 90.78 | 123.05 | 92.26 |
| Substance dependence | 152.68 | 2745.73 | 3428.49 | 3044.71 |
| Transcription | 839.55 | 7519.82 | 7282.92 | 9511.74 |
| Translation | 17,578.72 | 21,123.98 | 21,335.18 | 24,807.37 |
| Transport and catabolism | 1426.55 | 18,433.02 | 20,281.75 | 20,499.89 |
| Xenobiotics biodegradation and metabolism | 10,956.91 | 4275.83 | 3958.65 | 4369.27 |
| Category | A0 | A24 | A36 | A43 |
|---|---|---|---|---|
| GT:Glycosyl Transferases | 4560.21 | 4204.55 | 4017.57 | 4552.48 |
| GH:Glycoside Hydrolases | 2964.22 | 2396.13 | 2469.17 | 2358.73 |
| CBM:Carbohydrate-Binding Modules | 443.48 | 224.88 | 266.55 | 174.80 |
| CE:Carbohydrate Esterases | 274.82 | 80.65 | 77.281 | 66.236 |
| AA:AuxiliaryActivities | 56.36 | 6.05 | 4.01 | 2.29 |
| PL:Polysaccharide Lyases | 0.23 | 0 | 0 | 0.03 |
Appendix B
References
- Yue, F.; Hao, Z.; Jing, R.; Zhang, B. Research on the processing technology of new type fermented waxy Corn. Food RD FRD 2012, 33, 131–134. [Google Scholar]
- Yang, Y.; Chen, W.; Ma, R.; Xu, L. Analysis and evaluation of nutrients in sweet rice wine. China Brew. 2011, 30, 182–184. [Google Scholar]
- Ren, F.; Han, Z. Study on the fermentation mechanism of rice wine. China Brew. 2012, 31, 140–143. [Google Scholar]
- Tian, Y.; Wang, W.; Jia, C.; Liu, H. Study on fermentation process of sweetened glutinous rice wine and changes regularity of components during fermentation. Food Sci. Technol. 2008, 33, 37–39. [Google Scholar]
- Yang, Y.; Zhong, H.; Yang, N.; Zhu, D.; Li, J.; Yang, Z.; Yang, T. Effects of the proteins of indica rice and indica waxy rice on the formation of volatiles of sweet rice wine. Int. J. Food Sci. Technol. 2022, 57, 3604–3615. [Google Scholar]
- Zhong, J.; Ye, X.; Fang, Z.; Xie, G.; Liao, N.; Shu, J.; Liu, D. Determination of biogenic amines in semi-dry and semi-sweet Chinese rice wines from the Shaoxing region. Food Control 2012, 28, 151–156. [Google Scholar]
- Zheng, X.; Wang, H.; Qiao, X.; Cha, Y.; Gao, L. Development status of compound glutinous rice wine production technology. Winemaking 2023, 50, 26–32. [Google Scholar]
- Jeong, J.W.; Nam, P.W.; Lee, S.J.; Lee, K.G. Antioxidant activities of Korean rice wine concentrates. J. Agr. Food Chem. 2011, 59, 7039–7044. [Google Scholar]
- Peng, L.; Ai-Lati, A.; Ji, Z.; Chen, S.; Mao, J. Polyphenols extracted from huang jiu have anti-inflammatory activity in lipopolysaccharide stimulated RAW264. 7 cells. RSC Adv. 2019, 9, 5295–5301. [Google Scholar] [CrossRef]
- Lin, H.; Zhang, J.; Ni, T.; Lin, N.; Meng, L.; Gao, F.; Luo, H.; Liu, X.; Chi, J.; Guo, H. Yellow wine polyphenolic compounds prevents doxorubicin-induced cardiotoxicity through activation of the Nrf2 signalling pathway. J. Cell Mol. Med. 2019, 23, 6034–6047. [Google Scholar]
- Cai, H.; Zhang, T.; Zhang, Q.; Luo, J.; Cai, C.; Mao, J. Microbial diversity and chemical analysis of the starters used in traditional Chinese sweet rice wine. Food Microbiol. 2018, 73, 319–326. [Google Scholar] [PubMed]
- Gammacurta, M.; Lytra, G.; Marchal, A.; Marchand, S.; Barbe, J.C.; Moine, V.; de Revel, G. Influence of lactic acid bacteria strains on ester concentrations in red wines: Specific impact on branched hydroxylated compounds. Food Chem. 2018, 239, 252–259. [Google Scholar] [PubMed]
- Simonen, M.; Palva, I. Protein secretion in Bacillus species. Microbiol. Rev. 1993, 57, 109–137. [Google Scholar]
- Liu, Z.; Wang, Z.; Sun, J.; Ni, L. The dynamics of volatile compounds and their correlation with the microbial succession during the traditional solid-state fermentation of Gutian Hong Qu glutinous rice wine. Food Microbiol. 2020, 86, 103347. [Google Scholar]
- Lv, X.C.; Huang, Z.Q.; Zhang, W.; Rao, P.F.; Ni, L. Identification and characterization of filamentous fungi isolated from fermentation starters for Hong Qu glutinous rice wine brewing. J. Gen. Appl. Microbiol. 2012, 58, 33–42. [Google Scholar]
- Lv, X.C.; Cai, Q.Q.; Ke, X.X.; Chen, F.; Rao, P.F.; Ni, L. Characterization of fungal community and dynamics during the traditional brewing of Wuyi Hong Qu glutinous rice wine by means of multiple culture-independent methods. Food Control 2015, 54, 231–239. [Google Scholar]
- Yang, Y.; Zhong, H.; Yang, N.; Xu, S.; Yang, T. Quality improvement of sweet rice wine fermented with Rhizopus delemar on key aroma compounds content, phenolic composition, and antioxidant capacity compared to Rhizopus oryzae. J. Food Sci. Technol. 2022, 59, 2339–2350. [Google Scholar]
- Liang, Z.; Lin, X.; He, Z.; Su, H.; Li, W.; Ren, X. Amino acid and microbial community dynamics during the fermentation of Hong Qu glutinous rice wine. Food Microbiol. 2020, 90, 103467. [Google Scholar] [CrossRef]
- Su, Y.; Zhao, S.M. Research progress of process technology and quality characteristics of sweet rice wine. China Brew. 2014, 33, 27–30. [Google Scholar]
- Yang, Y.; Xia, Y.; Wang, G.; Yu, J.; Ai, L. Effect of mixed yeast starter on volatile flavor compounds in Chinese rice wine during different brewing stages. LWT 2017, 78, 373–381. [Google Scholar]
- Aleksandar, P.V.; Lisov, N.; Čakar, U.D.; Marković, N.; Matijašević, S.; Cvejić, J.M.; Atanacković, M.; Gojković-Bukarica, L. The effects of Prokupac variety clones and vinification method on the quantity of resveratrol in wine. Food Feed. Res. 2019, 46, 189–198. [Google Scholar]
- Zeng, Q.; Song, Y.; Liu, Z.; Xie, W.; Chen, Y.; Liang, S.; Yang, Y.; Liu, F.; Song, L. Research on the development and nutritional value of sweet wine Brewing. J. Foshan Univ. Sci. Technol. 2020, 38, 28–35. [Google Scholar]
- Xiong, J.; Xiang, Q.; Yan, X.; Wu, J. Changes in flavor, taste, and quality during the fermentation process of black glutinous rice sweet wine. China Brew. 2023, 42, 122–127. [Google Scholar]
- Lu, Y.; Peng, Y.; Zhang, Y.; Liu, S.; Cui, L. Optimization of process parameters for fermented sweet osmanthus wine. Agric. Prod. Process. 2021, 40–44. [Google Scholar] [CrossRef]
- Lv, X.C.; Weng, X.; Zhang, W.; Rao, P.F.; Ni, L. Microbial diversity of traditional fermentation starters for Hong Qu glutinous rice wine as determined by PCR-mediated DGGE. Food Control 2012, 28, 426–434. [Google Scholar]
- Tang, Q.; He, G.; Huang, J.; Wu, C.; Jin, Y.; Zhou, R. Characterizing relationship of microbial diversity and metabolite in Sichuan xiao qu. Front. Microbiol. 2019, 10, 696. [Google Scholar]
- Lv, X.C.; Jiang, Y.J.; Liu, J.; Guo, W.L.; Liu, Z.B.; Zhang, W.; Rao, P.F. Evaluation of different PCR primers for denaturing gradient gel electrophoresis (DGGE) analysis of fungal community structure in traditional fermentation starters used for Hong Qu glutinous rice wine. Int. J. Food Microbiol. 2017, 255, 58–65. [Google Scholar]
- Lv, X.C.; Chen, Z.C.; Jia, R.B.; Liu, Z.B.; Zhang, W.; Chen, S.J.; Rao, P.F.; Ni, L. Microbial community structure and dynamics during the traditional brewing of Fuzhou Hong Qu glutinous rice wine as determined by culture-dependent and culture-independent techniques. Food Control 2015, 57, 216–224. [Google Scholar]
- Liu, M.; Tang, Y.; Zhao, K.; Liu, Y.; Guo, X.; Ren, D.; Yao, W.; Tian, X.; Gu, Y.; Yi, B.; et al. Determination of the fungal community of pit mud in fermentation cellars for Chinese strong-flavor liquor, using DGGE and Illumina Mi Seq sequencing. Food Res. Int. 2017, 91, 80–87. [Google Scholar]
- Cocolin, L.; Campolongo, S.; Alessandria, V.; Dolci, P.; Rantsiou, K. Culture independent analyses and wine fermentation: An overview of achievements 10 years after first application. Ann. Microbiol. 2011, 61, 17–23. [Google Scholar]
- Prakitchaiwattana, C.J.; Fleet, G.H.; Heard, G.M. Application and evaluation of denaturing gradient gel electrophoresis to analyse the yeast ecology of wine grapes. FEMS Yeast Res. 2004, 4, 865–877. [Google Scholar] [PubMed]
- Liu, Z.; Li, J.; Wei, B.; Huang, T.; Xiao, Y.; Peng, Z.; Xie, M.; Xiong, T. Bacterial community and composition in Jiang-shui and Suan-cai revealed by high-throughput sequencing of 16S rRNA. Int. J. Food Microbiol. 2019, 306, 108271. [Google Scholar]
- Luo, F.; Yang, Z.; Zhong, K.; Huang, C.; Yu, Z.; Peng, Z.; Wu, Y.; Bu, Q.; Gao, H. Effects of Bacillus megaterium L222 on quality and bacterial diversity of Sichuan paocai. Food Res. Int. 2021, 140, 109994. [Google Scholar] [PubMed]
- Zhao, G.; Liu, C.; Hadiatullah, H.; Yao, Y.; Lu, F. Effect of Hericium erinaceus on bacterial diversity and volatile flavor changes of soy sauce. LWT 2021, 139, 110543. [Google Scholar]
- Zhang, Z.; Gao, Y.; Zhao, W.; Liu, X.; Zhang, H. Analysis of fungal dynamic changes in the natural fermentation broth of ‘Hongyang’ kiwifruit. PeerJ 2022, 10, e13286. [Google Scholar]
- Zhao, W.; Zhang, Z.; Gao, Y.; Liu, X.; Du, C.; Ma, F.; Wang, S.; Shi, W.; Yang, Y.; Zhang, H.; et al. Fungal dynamic changes in naturally fermented ‘Kyoho’ grape juice. Arch. Microbiol. 2022, 204, 556. [Google Scholar]
- Zou, J.; Chen, X.; Wang, C.; Liu, Y.; Li, M.; Pan, X.; Chang, X. Microbial communities and correlation between microbiota and volatile compounds in fermentation starters of Chinese sweet rice wine from different regions. Foods 2023, 12, 2932. [Google Scholar] [CrossRef]
- Sun, H.; Liu, G.; Zhao, W.; Bai, W.; Liang, J. Effects of different enzymes and yeast combinations on the quality of rice flavor Baijiu. China Brew. 2024, 43, 201–205. [Google Scholar]
- Souza-Coutinho, M.; Brasil, R.; Souza, C.; Sousa, P.; Malfeito-Ferreira, M. Consumers associate high-quality (fine) wines with complexity, persistence, and unpleasant emotional responses. Foods 2020, 9, 452. [Google Scholar]
- Böhmer, M.; Smoľak, D.; Ženišová, K.; Čaplová, Z.; Pangallo, D.; Puškárová, A.; Bučková, M.; Cabicarová, T.; Budiš, J.; Šoltýs, K.; et al. Comparison of microbial diversity during fermentation of two different wines. FEMS Microbiol. Lett. 2020, 367, fnaa150. [Google Scholar]
- Liu, A.; Yang, X.; Guo, Q.; Li, B.; Zheng, Y.; Shi, Y.; Zhu, L. Microbial communities and flavor compounds during the fermentation of traditional Hong Qu glutinous rice wine. Foods 2022, 11, 1097. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Zhu, S.; Wang, X.; Wang, X.; Liu, J.; Liu, S.; Liu, J.; Liu, Y.; Fan, G.; Duan, S. Application of intelligent sensory technology in engineering rice research. Food Ferment. Ind. 2015, 41, 251–254. [Google Scholar]
- Zhang, N.N.; Xu, Q.; Li, H.Y. Comparative study of DNA extraction methods for microbial genomes in fermented bean curd. China Brew. 2023, 42, 177–181. [Google Scholar] [CrossRef]
- Kumar, G.; Eble, J.E.; Gaither, M.R. A practical guide to sample preservation and pre-PCR processing of aquatic environmental DNA. Mol. Ecol. Resour. 2020, 20, 29–39. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Elolimy, A.; Alharthi, A.; Zeineldin, M.; Parys, C.; Loor, J. Residual feed intake divergence during the preweaning period is associated with unique hindgut microbiome and metabolome profiles in neonatal Holstein heifer calves. J. Anim. Sci. Biotechnol. 2020, 11, 13. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef]
- Steenwyk, J.L.; Rokas, A. Ggpubfigs: Colorblind-friendly color palettes and ggplot2 graphic system extensions for publication-quality scientific figures. Microbiol. Resour. Ann. 2021, 10, e0087121. [Google Scholar] [CrossRef]
- Chao, A.; Ricotta, C. Quantifying evenness and linking it to diversity, beta diversity, and similarity. Ecology 2019, 100, e02852. [Google Scholar] [CrossRef]
- Liu, K.; Zhang, Y.; Li, Q.; Li, H.; Long, D.; Yan, S.; Huang, W.; Long, R.; Huang, X. Ethnic differences shape the alpha but not beta diversity of gut microbiota from school children in the absence of environmental differences. Microorganisms 2020, 8, 254. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. App. Environ. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Ramette, A. Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 2007, 62, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Quantitative Ecology, 2nd ed.; Science Press: Beijing, China, 2011; Chapter 6; pp. 186–190. [Google Scholar]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 326–349. [Google Scholar]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Kristiansen, K.; Dahlgren, J.; Wang, J. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar]
- Qin, J.; Li, Y.; Cai, Z.; LI, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Coste, A.; Sousa, P.; Malfeito-Ferreira, M. Wine tasting based on emotional responses: An expedite approach to distinguish between warm and cool climate dry red wine styles. Food Res. Int. 2018, 106, 11–21. [Google Scholar] [CrossRef]
- Heo, J.; Kwak, H.S.; Kim, M.; Kim, J.H.; Baek, H.H.; Shin, H.; Lee, Y.; Lee, S.; Kim, S.S. Major sensory attributes and volatile compounds of Korean rice liquor (Yakju) affecting overall acceptance by young consumers. Foods 2020, 9, 722. [Google Scholar] [CrossRef]
- Xiao, Z.; Yu, D.; Niu, Y.; Ma, N.; Zhu, J. Characterization of different aroma-types of Chinese liquors based on their aroma profile by gas chromatography-mass spectrometry and sensory evaluation. Flavour. Fragr. J. 2016, 31, 217–227. [Google Scholar] [CrossRef]
- Chen, L.; Ren, L.; Li, D.; Ma, X. Analysis of microbiomes in three traditional starters and volatile components of the Chinese rice wines. Food Sci. Biotechnol. 2021, 30, 87–96. [Google Scholar] [PubMed]
- Lee, S.J.; Lee, K.G. Understanding consumer preferences for rice wines using sensory data. J. Sci. Food Agric. 2008, 88, 690–698. [Google Scholar]
- Wong, B.; Owens, A.; Phillips, M.; Kam, R. Identifying sensory attributes of Korean rice wine (makgeolli) using sensory evaluation and chemical analysis. J. Food Sci. 2023, 88, 4247–4261. [Google Scholar]
- Li, N.; Li, Y.; Sun, Y. Research progress on regulation mechanism of ethanol in Saccharomyces cerevisiae and breeding of low-yielding strains. Food Ferment. Ind. 2024, 50, 315–323. [Google Scholar]
- Wolfe, B.E.; Button, J.E.; Santarelli, M.; Dutton, R.J. Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity. Cell 2014, 158, 422–433.57. [Google Scholar]
- Huang, Z.R.; Guo, W.L.; Zhou, W.B.; Li, L.; Xu, J.X.; Hong, J.L.; Liu, H.P.; Zeng, F.; Bai, W.D.; Liu, B.; et al. Microbial communities and volatile metabolites in different traditional fermentation starters used for Hong Qu glutinous rice wine. Food Res. Int. 2019, 121, 593–603. [Google Scholar]
- Tian, S.; Zeng, W.; Zhou, J.; Du, G. Correlation between the microbial community and ethyl carbamate generated during Huzhou rice wine fermentation. Food Res. Int. 2022, 154, 111001. [Google Scholar]
- Fugelsang, K.C.; Edwards, C.G. Chapter: Fermentation and Post-Fermentation Processing. In Wine Microbiology: Practical Applications and Procedures; Springer US: Boston, MA, USA, 2007; pp. 115–138. [Google Scholar]
- Fleet, G.H. The microorganisms of winemaking-isolation, enumeration and identification. In Wine Microbiology and Biotechnology; CRC Press: Boca Raton, FL, USA, 1993; pp. 1–25. [Google Scholar]
- Padilla, B.; Gil, J.V.; Manzanares, P. Challenges of the non-conventional yeast Wickerhamomyces anomalus in winemaking. Fermentation 2018, 4, 68. [Google Scholar] [CrossRef]
- Sabel, A.; Martens, S.; Petri, A.; König, H.; Claus, H. Wickerhamomyces anomalus AS1: A new strain with potential to improve wine aroma. Ann. Microbiol. 2014, 64, 483–491. [Google Scholar]
- Sun, N.; Gao, Z.; Li, S.; Chen, X.; Guo, J. Assessment of chemical constitution and aroma properties of Kiwi wines obtained from pure and mixed fermentation with Wickerhamomyces anomalus and Saccharomyces cerevisiae. J. Sci. Food Agric. 2022, 102, 175–184. [Google Scholar]
- Englezos, V.; Jolly, N.P.; Di Gianvito, P.; Rantsiou, K.; Cocolin, L. Microbial interactions in winemaking: Ecological aspects and effect on wine quality. Trends Food Sci. Tech. 2022, 127, 99–113. [Google Scholar]
- Wu, J.; Ren, L.; Zhao, N.; Wu, T.; Liu, R.; Sui, W.; Zhang, M. Solid-state fermentation by Rhizopus oryzae improves flavor of wheat bran for application in food. J. Cereal Sci. 2022, 107, 103536. [Google Scholar]
- Wang, L.; Wang, Y.; Wang, H.; Liu, G.; Yang, F.; Jiang, H.; Xu, J.; Wang, D.; Jin, T. Microbial composition analysis of pit mud of Maotai flavor Baijiu. Brew. Technol. 2015, 12–15. [Google Scholar] [CrossRef]
- Li, H.; Wang, H.; Wu, Y.; Huang, W. Isolation, identification, and evaluation of brewing performance of natural wine yeast strains. Food Ferment. Ind. 2010, 36, 14–20. [Google Scholar]
- Lu, J.; San, Q.M.G.; Tang, P.; Wang, L.; Meng, T.; Liang, Q.; Zhang, L.; Feng, G.; Li, C. Screening of acid resistant yeast strains and their application in the brewing of Maotai flavor Baijiu. Brew. Technol. 2019, 106–111. [Google Scholar]
- Cheng, J.; Guo, W.; Hu, H. Isolation and performance determination of an acid resistant Yeast strain. China Brew. 2013, 32, 39–42. [Google Scholar]
- Wang, Z.; Zhang, Y.; Meng, Q.; Chen, X.; Qi, H.; Li, H.; Yan, Z. Screening of high-temperature resistant and high-yield alcohol producing yeast and its application in Daqu production. Brew. Technol. 2020, 107–114. [Google Scholar] [CrossRef]
- Li, H.; Li, Z.; Qu, J.; Wang, J. Bacterial diversity in traditional Jiaozi and sourdough revealed by high-throughput sequencing of 16S rRNA amplicons. LWT 2017, 81, 319–325. [Google Scholar]
- Xiao, C.; Wang, L.; Zhang, Y.G.; Tu, T.Y.; Wang, S.T.; Shen, C.H.; Yuan, H.W.; Zhong, X.Z. A comparison of microbial communities and volatile compounds in wheat Qu from different geographic locations. LWT 2021, 148, 111752. [Google Scholar]
- Zhao, X.; Xiang, F.; Tang, F.; Cai, W.; Guo, Z.; Hou, Q.; Yang, X.; Song, W.; Shan, C. Bacterial communities and prediction of microbial metabolic pathway in rice wine Koji from different regions in China. Front. Microbiol. 2022, 12, 748779. [Google Scholar]
- Yang, D.; Liu, L.; Li, P. Analysis of correlation between acetyl coenzyme A and Saccharomyces cerevisiae growth, esters production in beer fermentation under top CO2 pressure. China Brew. 2019, 38, 137–141. [Google Scholar]
| Project | Scoring Criteria | Full Marks |
|---|---|---|
| Aroma | No aroma or peculiar smell, almost no wine aroma. (0~4) With aroma, no peculiar smell, and insufficient wine aroma. (5~7) Strong aroma, with glutinous rice aroma and light wine aroma. (8~10) | 10 |
| Texture | The color is dark or has a different color, and almost no wine seepage. (0~4) The color is uneven, the gloss is insufficient, and the amount of wine seepage is small. (5~7) The color is uniform, the gloss is good, and the wine seepage is normal. (8~10) | 10 |
| Taste | Poor taste, poor elasticity, undercooked or soft. (0~4) Average taste, insufficient elasticity, soft or hard. (5~7) Delicious taste, good elasticity, moderate hardness. (8~10) | 10 |
| Fermentation Cycle (h) | 0 | 24 | 36 | 43 | |
|---|---|---|---|---|---|
| Physical and chemical indicators | pH | 6.19 ± 0.001 | 3.86 ± 0.012 | 3.89 ± 0.010 | 3.75 ± 0.012 |
| Total acid (g/100 g SFR) | Not detected | 0.17 ± 0.069 | 0.24 ± 0.054 | 0.34 ± 0.032 | |
| Total sugar (°Brix) | Not detected | 10 ± 0.1 | 21 ± 0.1 | 33 ± 0.0 | |
| Alcohol content (% v/v) | Not detected | Not detected | Not detected | 3.50 ± 0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, J.; Tian, P.; Liu, X.; Zhang, H. Analysis of the Changes in Physicochemical Properties and Microbial Communities During Fermentation of Sweet Fermented Rice. Foods 2025, 14, 1121. https://doi.org/10.3390/foods14071121
Wan J, Tian P, Liu X, Zhang H. Analysis of the Changes in Physicochemical Properties and Microbial Communities During Fermentation of Sweet Fermented Rice. Foods. 2025; 14(7):1121. https://doi.org/10.3390/foods14071121
Chicago/Turabian StyleWan, Jiaqiong, Ping Tian, Xiaozhen Liu, and Hanyao Zhang. 2025. "Analysis of the Changes in Physicochemical Properties and Microbial Communities During Fermentation of Sweet Fermented Rice" Foods 14, no. 7: 1121. https://doi.org/10.3390/foods14071121
APA StyleWan, J., Tian, P., Liu, X., & Zhang, H. (2025). Analysis of the Changes in Physicochemical Properties and Microbial Communities During Fermentation of Sweet Fermented Rice. Foods, 14(7), 1121. https://doi.org/10.3390/foods14071121