
Quantitative Analysis of Pyrrolizidine Alkaloids in Food Matrices and Plant-Derived Samples Using UHPLC—MS/MS

 , ,
, ,
Abstract
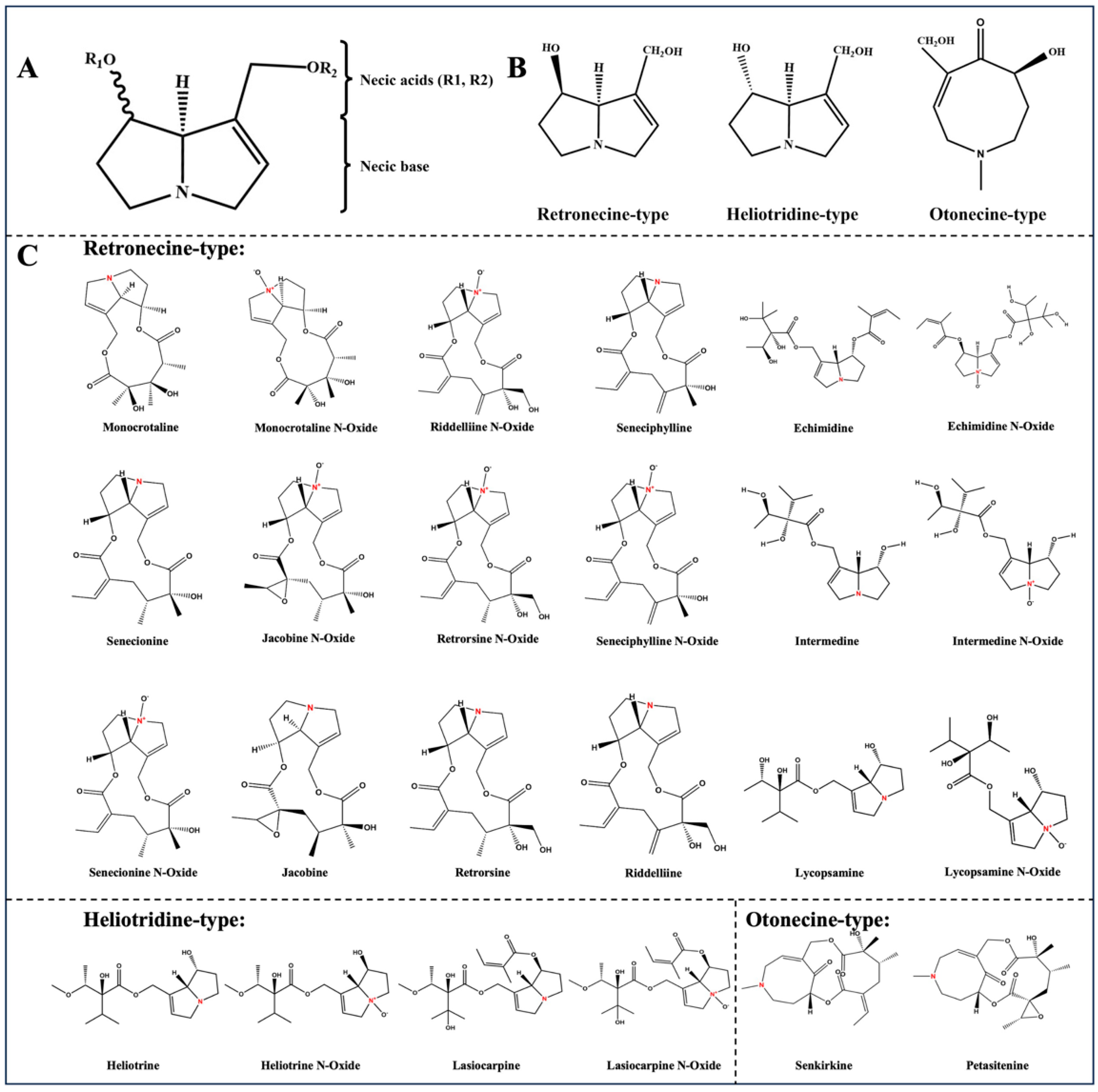
:1. Introduction
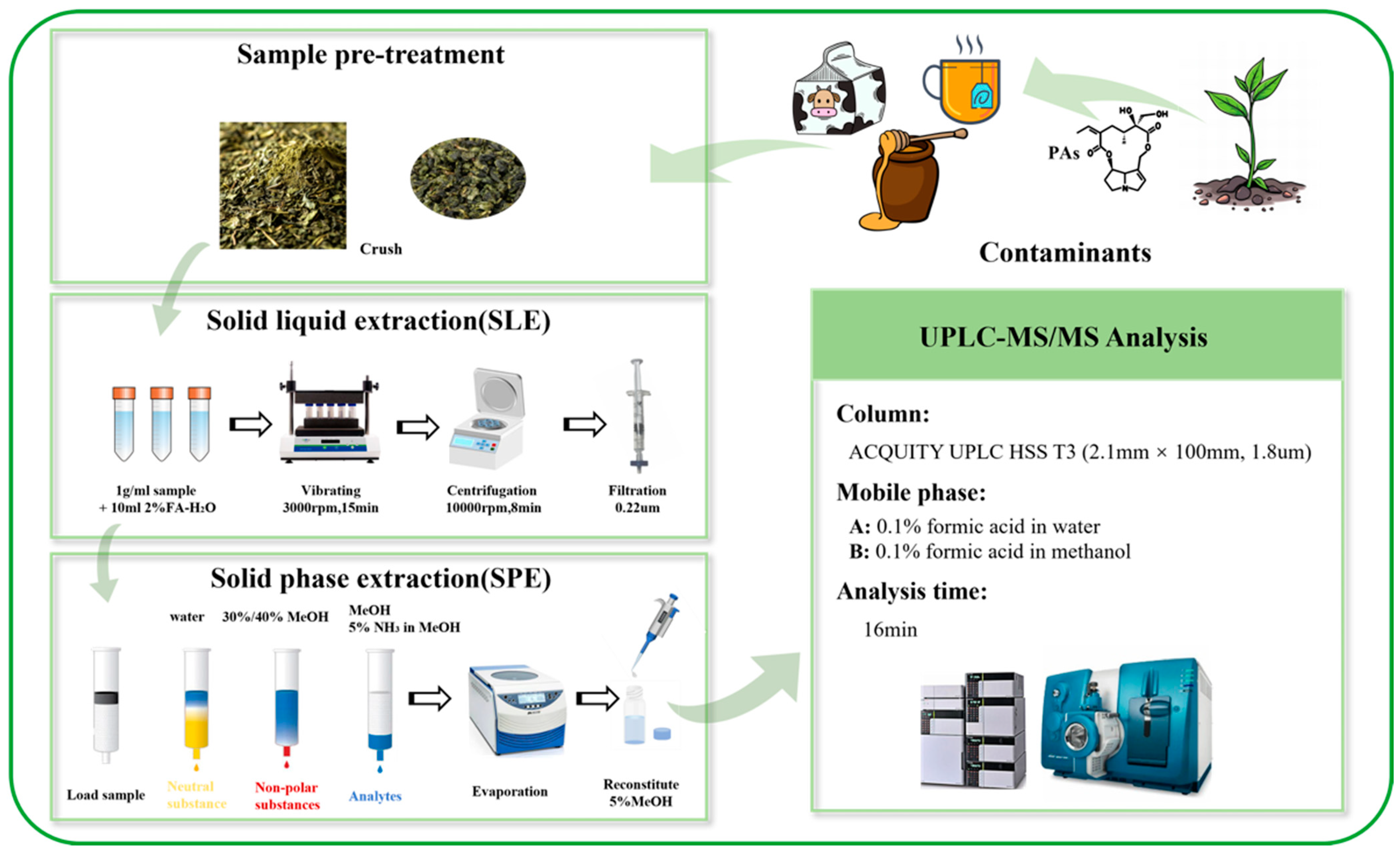
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Sample Collection
2.3. Sample Preparation
2.4. Preparation of the Matrix-Matched Calibration Standards
2.5. HPLC Analysis
2.6. Method Validation
3. Results and Discussion
3.1. Optimization of the Chromatographic Separation Conditions for the 24 PAs
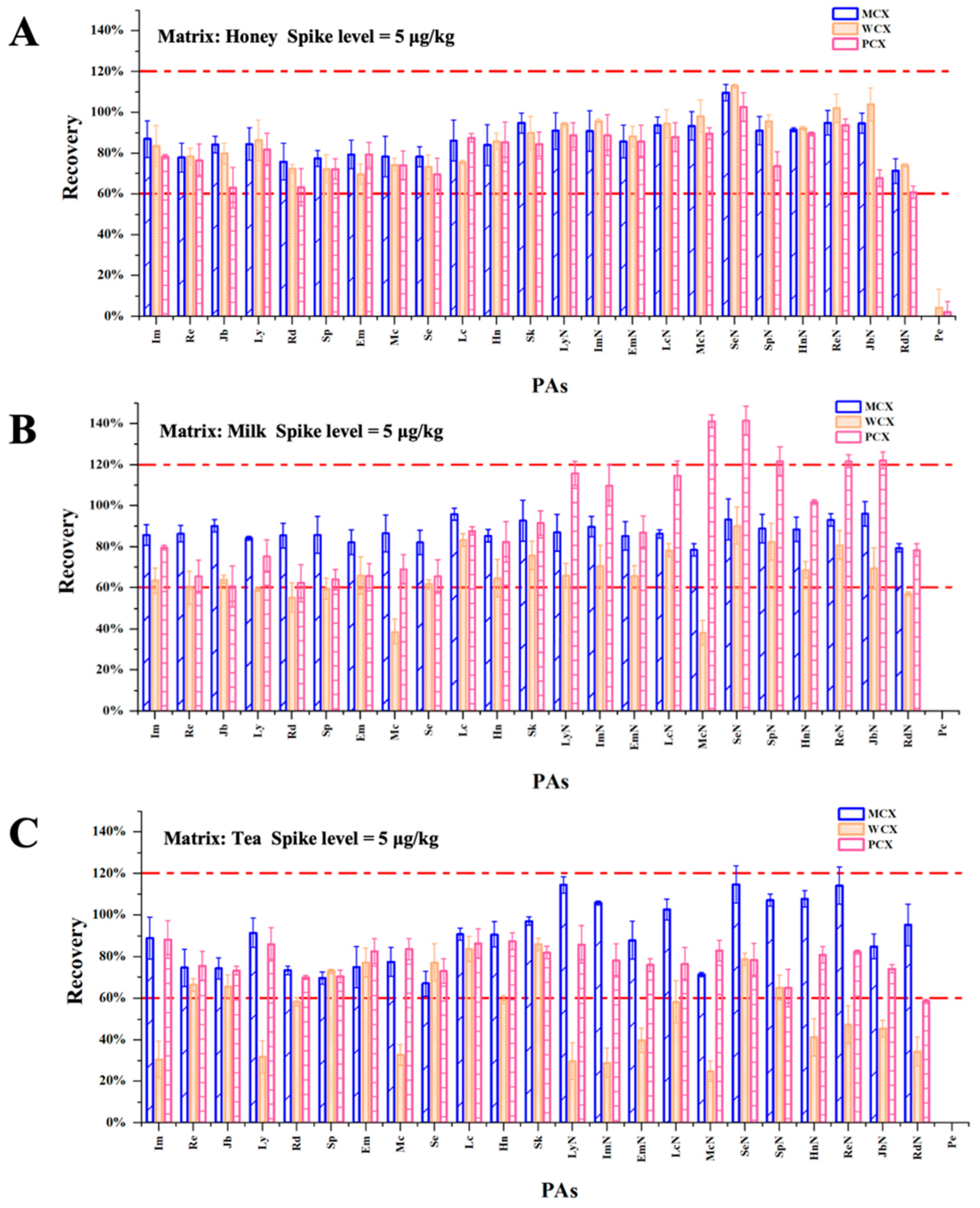
3.2. Selection of the SPE Conditions
3.3. Optimization of the Rinsing Solution and Selection of the Extraction Solvent
3.4. Method Validation
3.4.1. Linearity, LODs and LOQs
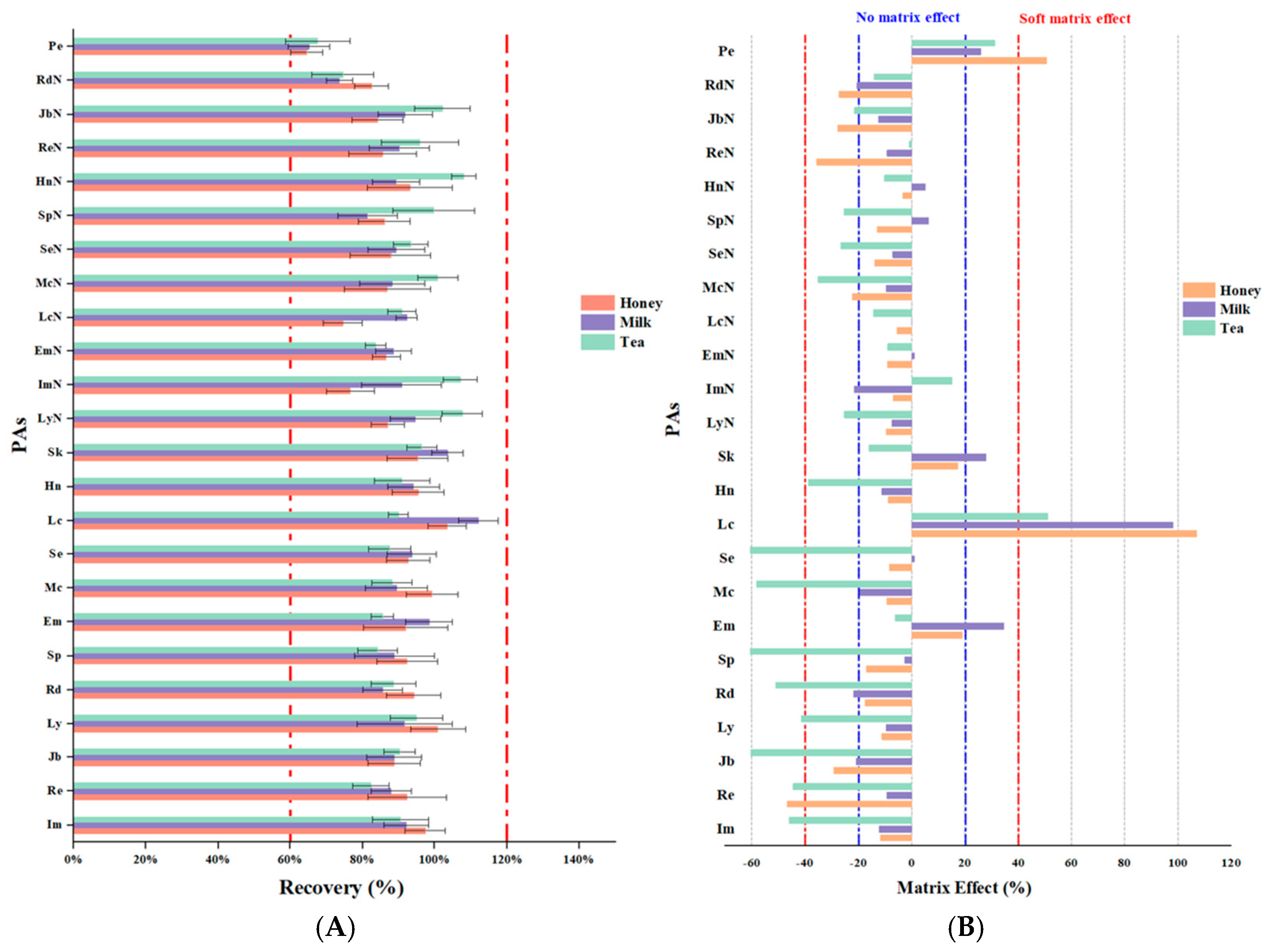
3.4.2. Recovery and Precision
3.4.3. Evaluation of the Matrix Effect
3.5. Comparison with Other Methods
3.6. Analysis of Commercial Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louisse, J.; Mulder, P.P.J.; Gerssen, A.; Stoopen, G.; Rijkers, D.; van de Schans, M.G.M.; Peijnenburg, A. Bioassay-directed analysis-based identification of relevant pyrrolizidine alkaloids. Arch. Toxicol. 2022, 96, 2299–2317. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, E.; Kwiatek, K. Simultaneous Determination of Pyrrolizidine and Tropane Alkaloids in Honey by Liquid Chromatography-mass Spectrometry. J. Vet. Res. 2022, 66, 235–243. [Google Scholar] [CrossRef]
- Martinello, M.; Borin, A.; Stella, R.; Bovo, D.; Biancotto, G.; Gallina, A.; Mutinelli, F. Development and validation of a QuEChERS method coupled to liquid chromatography and high resolution mass spectrometry to determine pyrrolizidine and tropane alkaloids in honey. Food Chem. 2017, 234, 295–302. [Google Scholar] [CrossRef]
- Lin, G.; Wang, J.Y.; Li, N.; Li, M.; Gao, H.; Ji, Y.; Zhang, F.; Wang, H.; Zhou, Y.; Ye, Y.; et al. Hepatic sinusoidal obstruction syndrome associated with consumption of Gynura segetum. J. Hepatol. 2011, 54, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Enge, A.M.; Kaltner, F.; Gottschalk, C.; Braeuning, A.; Hessel-Pras, S. Active Transport of Hepatotoxic Pyrrolizidine Alkaloids in HepaRG Cells. Int. J. Mol. Sci. 2021, 22, 3821. [Google Scholar] [CrossRef]
- Lu, Y.; Wong, K.Y.; Tan, C.; Ma, J.; Feng, B.; Lin, G. Establishment of a novel CYP3A4-transduced human hepatic sinusoidal endothelial cell model and its application in screening hepatotoxicity of pyrrolizidine alkaloids. J. Environ. Sci. Health C Toxicol. Carcinog. 2020, 38, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Gao, H.; Li, N.; Xue, J.; Chen, J.; Ke, C.; Ye, Y.; Fu, P.P.; Zheng, J.; Wang, J.; et al. Blood Pyrrole-Protein Adducts—A Biomarker of Pyrrolizidine Alkaloid-Induced Liver Injury in Humans. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2015, 33, 404–421. [Google Scholar] [CrossRef]
- Schrenk, D.; Gao, L.; Lin, G.; Mahony, C.; Mulder, P.P.J.; Peijnenburg, A.; Pfuhler, S.; Rietjens, I.; Rutz, L.; Steinhoff, B.; et al. Pyrrolizidine alkaloids in food and phytomedicine: Occurrence, exposure, toxicity, mechanisms, and risk assessment—A review. Food Chem. Toxicol. 2020, 136, 111107. [Google Scholar] [CrossRef]
- Zhu, L.; Xue, J.; He, Y.; Xia, Q.; Fu, P.P.; Lin, G. Correlation Investigation between Pyrrole-DNA and Pyrrole-Protein Adducts in Male ICR Mice Exposed to Retrorsine, a Hepatotoxic Pyrrolizidine Alkaloid. Toxins 2022, 14, 377. [Google Scholar] [CrossRef]
- Avula, B.; Sagi, S.; Wang, Y.H.; Zweigenbaum, J.; Wang, M.; Khan, I.A. Characterization and screening of pyrrolizidine alkaloids and N-oxides from botanicals and dietary supplements using UHPLC-high resolution mass spectrometry. Food Chem. 2015, 178, 136–148. [Google Scholar] [CrossRef]
- Madge, I.; Cramer, L.; Rahaus, I.; Jerz, G.; Winterhalter, P.; Beuerle, T. Pyrrolizidine alkaloids in herbal teas for infants, pregnant or lactating women. Food Chem. 2015, 187, 491–498. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xia, Q.; Shi, Q.; Fu, P.P. Metabolism of carcinogenic pyrrolizidine alkaloids and pyrrolizidine alkaloid N-oxides by rat primary hepatocytes generate the same characteristic DHP-DNA adducts. J. Environ. Sci. Health C Toxicol. Carcinog. 2021, 39, 357–372. [Google Scholar] [CrossRef]
- He, Y.; Zhu, L.; Ma, J.; Lin, G. Metabolism-mediated cytotoxicity and genotoxicity of pyrrolizidine alkaloids. Arch. Toxicol. 2021, 95, 1917–1942. [Google Scholar] [CrossRef] [PubMed]
- Kempf, M.; Reinhard, A.; Beuerle, T. Pyrrolizidine alkaloids (PAs) in honey and pollen-legal regulation of PA levels in food and animal feed required. Mol. Nutr. Food Res. 2010, 54, 158–168. [Google Scholar] [CrossRef] [PubMed]
- de Nijs, M.; Mulder, P.P.J.; Klijnstra, M.D.; Driehuis, F.; Hoogenboom, R. Fate of pyrrolizidine alkaloids during processing of milk of cows treated with ragwort. Food Addit. Contam. Part. A Chem. Anal. Control Expo. Risk Assess. 2017, 34, 2212–2219. [Google Scholar] [CrossRef]
- Reinhard, H.; Zoller, O. Pyrrolizidine alkaloids in tea, herbal tea and iced tea beverages- survey and transfer rates. Food Addit. Contam. Part. A Chem. Anal. Control Expo. Risk Assess. 2021, 38, 1914–1933. [Google Scholar] [CrossRef]
- Kucukoglu, A.S.; Hiz, G.; Karaca, H. Effects of thermal and nonthermal treatments on microorganisms, pyrrolizidine alkaloids and volatile compounds in oregano (Origanum vulgare L.). Food Chem. 2024, 440, 138235. [Google Scholar] [CrossRef]
- De Jesus Inacio, L.; Merlanti, R.; Lucatello, L.; Bisutti, V.; Carraro, L.; Larini, I.; Vitulo, N.; Cardazzo, B.; Capolongo, F. Natural contaminants in bee pollen: DNA metabarcoding as a tool to identify floral sources of pyrrolizidine alkaloids and fungal diversity. Food Res. Int. 2021, 146, 110438. [Google Scholar] [CrossRef]
- Hoogenboom, L.A.; Mulder, P.P.; Zeilmaker, M.J.; van den Top, H.J.; Remmelink, G.J.; Brandon, E.F.; Klijnstra, M.; Meijer, G.A.; Schothorst, R.; Van Egmond, H.P. Carry-over of pyrrolizidine alkaloids from feed to milk in dairy cows. Food Addit. Contam. Part. A Chem. Anal. Control Expo. Risk Assess. 2011, 28, 359–372. [Google Scholar] [CrossRef]
- Huybrechts, B.; Callebaut, A. Pyrrolizidine alkaloids in food and feed on the Belgian market. Food Addit. Contam. Part. A Chem. Anal. Control Expo. Risk Assess. 2015, 32, 1939–1951. [Google Scholar] [CrossRef]
- Van Wyk, B.E.; Stander, M.A.; Long, H.S. Senecio angustifolius as the major source of pyrrolizidine alkaloid contamination of rooibos tea (Aspalathus linearis). S. Afr. J. Bot. 2017, 110, 124–131. [Google Scholar] [CrossRef]
- Zhu, L.; Zhang, C.Y.; Li, D.P.; Chen, H.B.; Ma, J.; Gao, H.; Ye, Y.; Wang, J.Y.; Fu, P.P.; Lin, G. Tu-San-Qi (Gynura japonica): The culprit behind pyrrolizidine alkaloid-induced liver injury in China. Acta Pharmacol. Sin. 2021, 42, 1212–1222. [Google Scholar] [CrossRef]
- Chain, E.P.o.C.i.t.F.; Knutsen, H.K.; Alexander, J.; Barregard, L.; Bignami, M.; Bruschweiler, B.; Ceccatelli, S.; Cottrill, B.; Dinovi, M.; Edler, L.; et al. Risks for human health related to the presence of pyrrolizidine alkaloids in honey, tea, herbal infusions and food supplements. EFSA J. 2017, 15, e04908. [Google Scholar] [CrossRef]
- Bundesinstitut für Risikobewertung (BfR). Pyrrolizidine Alkaloids in Herbal Teas and Teas; Bundesinstitut für Risikobewertung: Berlin, Germany, 2013. [Google Scholar]
- European Commission. Commission Regulation (EU) 2020/2040 of 11 December 2020 Amending Regulation (EC) No 1881/2006 as Regards Maximum Levels of Pyrrolizidine Alkaloids in Certain Foodstuffs (Text with EEA Relevance). Off. J. Eur. Union 2020, 63, 1–5. Available online: https://eur-lex.europa.eu/eli/reg/2020/2040/oj (accessed on 17 March 2025).
- Moreira, R.; Fernandes, F.; Valentao, P.; Pereira, D.M.; Andrade, P.B. Echium plantagineum L. honey: Search of pyrrolizidine alkaloids and polyphenols, anti-inflammatory potential and cytotoxicity. Food Chem. 2020, 328, 127169. [Google Scholar] [CrossRef]
- Chen, Y.; Li, L.; Xiong, F.; Xie, Y.; Xiong, A.; Wang, Z.; Yang, L. Rapid identification and determination of pyrrolizidine alkaloids in herbal and food samples via direct analysis in real-time mass spectrometry. Food Chem. 2021, 334, 127472. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Schoch, T.K.; Stegelmeier, B.L.; Gardner, D.R.; Than, K.A.; Molyneux, R.J. Development of enzyme-linked immunosor-bent assays for the hepatotoxic alkaloids riddelliine and riddelliine N-oxide. J. Agric. Food Chem. 2001, 49, 4144–4151. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Li, S.F. Dynamic pH junction-sweeping capillary electrophoresis for online preconcentration of toxic pyrrolizidine alkaloids in Chinese herbal medicine. Electrophoresis 2005, 26, 4360–4367. [Google Scholar] [CrossRef]
- Kowalczyk, E.; Sieradzki, Z.; Kwiatek, K. Determination of Pyrrolizidine Alkaloids in Honey with Sensitive Gas Chromatography-Mass Spectrometry Method. Food Anal. Methods 2017, 11, 1345–1355. [Google Scholar] [CrossRef]
- De Jesus Inacio, L.; Merlanti, R.; Lucatello, L.; Bisutti, V.; Contiero, B.; Serva, L.; Segato, S.; Capolongo, F. Pyrrolizidine alkaloids in bee pollen identified by LC-MS/MS analysis and colour parameters using multivariate class modeling. Heliyon 2020, 6, e03593. [Google Scholar] [CrossRef]
- Garcia-Juan, A.; Leon, N.; Armenta, S.; Pardo, O. Development and validation of an analytical method for the simultaneous determination of 12 ergot, 2 tropane, and 28 pyrrolizidine alkaloids in cereal-based food by LC-MS/MS. Food Res. Int. 2023, 174, 113614. [Google Scholar] [CrossRef] [PubMed]
- Gunthardt, B.F.; Wettstein, F.E.; Hollender, J.; Singer, H.; Harri, J.; Scheringer, M.; Hungerbuhler, K.; Bucheli, T.D. Retrospective HRMS Screening and Dedicated Target Analysis Reveal a Wide Exposure to Pyrrolizidine Alkaloids in Small Streams. Environ. Sci. Technol. 2021, 55, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wan, S.Y.; Jiang, Z.; Li, S.F.; Ong, E.S.; Osorio, J.C. Determination of pyrrolizidine alkaloids in comfrey by liquid chromatography-electrospray ionization mass spectrometry. Talanta 2009, 80, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Liu, Y.; Zhu, L.; Ji, H.; Song, X.; Guo, H.; Yi, T. Determination and regulation of hepatotoxic pyrrolizidine alkaloids in food: A critical review of recent research. Food Chem. Toxicol. 2018, 119, 50–60. [Google Scholar] [CrossRef]
- Sattler, M.; Muller, V.; Bunzel, D.; Kulling, S.E.; Soukup, S.T. Pyrrolizidine alkaloids in borage (Borago officinalis): Comprehensive profiling and development of a validated LC-MS/MS method for quantification. Talanta 2023, 258, 124425. [Google Scholar] [CrossRef]
- Urban, M.; Hann, S.; Rost, H. Simultaneous determination of pesticides, mycotoxins, tropane alkaloids, growth regulators, and pyrrolizidine alkaloids in oats and whole wheat grains after online clean-up via two-dimensional liquid chromatography tandem mass spectrometry. J. Environ. Sci. Health B 2019, 54, 98–111. [Google Scholar] [CrossRef]
- Yoon, S.H.; Kim, M.S.; Kim, S.H.; Park, H.M.; Pyo, H.; Lee, Y.M.; Lee, K.T.; Hong, J. Effective application of freezing lipid precipitation and SCX-SPE for determination of pyrrolizidine alkaloids in high lipid foodstuffs by LC-ESI-MS/MS. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 2015, 992, 56–66. [Google Scholar] [CrossRef]
- Izcara, S.; Casado, N.; Morante-Zarcero, S.; Perez-Quintanilla, D.; Sierra, I. Miniaturized and modified QuEChERS method with mesostructured silica as clean-up sorbent for pyrrolizidine alkaloids determination in aromatic herbs. Food Chem. 2022, 380, 132189. [Google Scholar] [CrossRef]
- Jansons, M.; Fedorenko, D.; Pavlenko, R.; Berzina, Z.; Bartkevics, V. Nanoflow liquid chromatography mass spectrometry method for quantitative analysis and target ion screening of pyrrolizidine alkaloids in honey, tea, herbal tinctures, and milk. J. Chromatogr. A 2022, 1676, 463269. [Google Scholar] [CrossRef]
- Wu, H.; Fan, D.; Cheng, J. Development and Validation of an UHPLC-MS/MS Method for the Determination of 32 Pyrrolizidine Alkaloids in Chinese Wild Honey. J. AOAC Int. 2022, 106, 56–64. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Q.; Yi, Z.; Chen, Y.; Xiao, W.; Su, D.; Shi, W. Risk Assessment of (Herbal) Teas Containing Pyrrolizidine Alkaloids (PAs) Based on Margin of Exposure Approach and Relative Potency (REP) Factors. Foods 2022, 11, 2946. [Google Scholar] [CrossRef]
- Peloso, M.; Minkoumba Sonfack, G.; Paduano, S.; De Martino, M.; De Santis, B.; Caprai, E. Pyrrolizidine Alkaloids in Food on the Italian Market. Molecules 2023, 28, 5346. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, Z.; Wong, L.; He, Y.; Zhao, Z.; Ye, Y.; Fu, P.P.; Lin, G. Contamination of hepatotoxic pyrrolizidine alkaloids in retail honey in China. Food Control 2018, 85, 484–494. [Google Scholar] [CrossRef]
- Buchmueller, J.; Sprenger, H.; Ebmeyer, J.; Rasinger, J.D.; Creutzenberg, O.; Schaudien, D.; Hengstler, J.G.; Guenther, G.; Braeuning, A.; Hessel-Pras, S. Pyrrolizidine alkaloid-induced transcriptomic changes in rat lungs in a 28-day subacute feeding study. Arch. Toxicol. 2021, 95, 2785–2796. [Google Scholar] [CrossRef] [PubMed]
- Castells, E.; Mulder, P.P.; Perez-Trujillo, M. Diversity of pyrrolizidine alkaloids in native and invasive Senecio pterophorus (Asteraceae): Implications for toxicity. Phytochemistry 2014, 108, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Kisielius, V.; Hama, J.R.; Skrbic, N.; Hansen, H.C.B.; Strobel, B.W.; Rasmussen, L.H. The invasive butterbur contaminates stream and seepage water in groundwater wells with toxic pyrrolizidine alkaloids. Sci. Rep. 2020, 10, 19784. [Google Scholar] [CrossRef]
- Klevenhusen, F.; These, A.; Taenzer, J.; Weiss, K.; Pieper, R. Effects of ensiling conditions on pyrrolizidine alkaloid degradation in silages mixed with two different Senecio spp. Arch. Anim. Nutr. 2022, 76, 93–111. [Google Scholar] [CrossRef]
- Schultze, A.E.; Roth, R.A. Chronic pulmonary hypertension—The monocrotaline model and involvement of the hemostatic system. J. Toxicol. Environ. Health B Crit. Rev. 1998, 1, 271–346. [Google Scholar] [CrossRef]
- FDA. Bioanalytical Method Validation, Guidance for Industry; US Food and Drug Administration. May 2018. Available online: https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm.pdf (accessed on 17 March 2025).
- Jiao, W.; Zhu, L.; Shen, T.; Wang, L.; Li, Q.X.; Wang, C.; Wu, X.; Chen, H.; Hua, R. Simultaneous determination of 15 pyrrolizidine alkaloids and their N-oxides in weeds, soil, fresh tea leaves, and tea: Exploring the pollution source of pyrrolizidine alkaloids in tea. Food Chem. 2024, 434, 137305. [Google Scholar] [CrossRef]
- Friedle, C.; Kapp, T.; Wallner, K.; Alkattea, R.; Vetter, W. High abundance of pyrrolizidine alkaloids in bee pollen collected in July 2019 from Southern Germany. Environ Monit Assess 2022, 194, 250. [Google Scholar] [CrossRef]
- Tsiokanos, E.; Tsafantakis, N.; Obe, H.; Beuerle, T.; Leti, M.; Fokialakis, N.; Grondin, A. Profiling of pyrrolizidine alkaloids using a retronecine-based untargeted metabolomics approach coupled to the quantitation of the retronecine-core in medicinal plants using UHPLC-QTOF. J. Pharm. Biomed. Anal. 2023, 224, 115171. [Google Scholar] [CrossRef] [PubMed]
- Hama, J.R.; Strobel, B.W. Pyrrolizidine alkaloids quantified in soil and water using UPLC-MS/MS. RSC Adv. 2019, 9, 30350–30357. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Jiang, C.; Wang, C.; Lu, Y.; Wang, Z.; Chai, Y.; Zhang, X.; Liu, X.; Lu, C.; Chen, H. Dissipation pattern and conversion of pyrrolizidine alkaloids (PAs) and pyrrolizidine alkaloid N-oxides (PANOs) during tea manufacturing and brewing. Food Chem. 2022, 390, 133183. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, E.; Kwiatek, K. Pyrrolizidine Alkaloids in Honey: Determination with Liquid Chromatography-mass Spectrometry Method. J. Vet. Res. 2018, 62, 173–181. [Google Scholar] [CrossRef]
- Chen, L.H.; Wang, J.C.; Guo, Q.L.; Qiao, Y.; Wang, H.J.; Liao, Y.H.; Sun, D.A.; Si, J.Y. Simultaneous Determination and Risk Assessment of Pyrrolizidine Alkaloids in Artemisia capillaris Thunb. by UPLC-MS/MS Together with Chemometrics. Molecules 2019, 24, 1077. [Google Scholar] [CrossRef]
- Jeong, S.H.; Choi, E.Y.; Kim, J.; Lee, C.; Kang, J.; Cho, S.; Ko, K.Y. LC-ESI-MS/MS Simultaneous Analysis Method Coupled with Cation-Exchange Solid-Phase Extraction for Determination of Pyrrolizidine Alkaloids on Five Kinds of Herbal Medicines. J. AOAC Int. 2021, 104, 1514–1525. [Google Scholar] [CrossRef]
- Bodi, D.; Ronczka, S.; Gottschalk, C.; Behr, N.; Skibba, A.; Wagner, M.; Lahrssen-Wiederholt, M.; Preiss-Weigert, A.; These, A. Determination of pyrrolizidine alkaloids in tea, herbal drugs and honey. Food Addit. Contam. Part. A Chem. Anal. Control Expo. Risk Assess. 2014, 31, 1886–1895. [Google Scholar] [CrossRef]
- Rizzo, S.; Celano, R.; Piccinelli, A.L.; Russo, M.; Rastrelli, L. Target screening method for the quantitative determination of 118 pyrrolizidine alkaloids in food supplements, herbal infusions, honey and teas by liquid chromatography coupled to quadrupole orbitrap mass spectrometry. Food Chem. 2023, 423, 136306. [Google Scholar] [CrossRef]
- Valese, A.C.; Daguer, H.; Muller, C.M.O.; Molognoni, L.; da Luz, C.F.P.; de Barcellos Falkenberg, D.; Gonzaga, L.V.; Brugnerotto, P.; Gorniak, S.L.; Barreto, F.; et al. Quantification of pyrrolizidine alkaloids in Senecio brasiliensis, beehive pollen, and honey by LC-MS/MS. J. Environ. Sci. Health B 2021, 56, 685–694. [Google Scholar] [CrossRef]
- Bolechova, M.; Caslavsky, J.; Pospichalova, M.; Kosubova, P. UPLC-MS/MS method for determination of selected pyrrolizidine alkaloids in feed. Food Chem. 2015, 170, 265–270. [Google Scholar] [CrossRef]
- Kwon, Y.; Koo, Y.; Jeong, Y. Determination of Pyrrolizidine Alkaloids in Teas Using Liquid Chromatography-Tandem Mass Spectrometry Combined with Rapid-Easy Extraction. Foods 2021, 10, 2250. [Google Scholar] [CrossRef] [PubMed]
- Knoop, K.; Klein, L.M.; Knispel, A.M.; Kaltner, F.; Gottschalk, C.; Knappstein, K.; Saltzmann, J.; Danicke, S. Dose-response study on the transfer of pyrrolizidine alkaloids from a tansy ragwort extract (Jacobaea vulgaris Gaertn.) to bovine milk. Food Addit. Contam. Part. A Chem. Anal. Control Expo. Risk Assess. 2024, 41, 1144–1157. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.M.; Gabler, A.M.; Rychlik, M.; Gottschalk, C.; Kaltner, F. A sensitive LC-MS/MS method for isomer separation and quantitative determination of 51 pyrrolizidine alkaloids and two tropane alkaloids in cow’s milk. Anal. Bioanal. Chem. 2022, 414, 8107–8124. [Google Scholar] [CrossRef] [PubMed]
- Girard, M.F.C.; Knight, P.; Hopfgartner, G. Vacuum differential mobility spectrometry combined with column-switching liquid chromatography- mass spectrometry for the analysis of pyrrolizidine alkaloids in tea samples. J. Chromatogr. A 2023, 1705, 464174. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Retention Time (min) | Precursor Ion [m/z] | Product Ions [m/z] (QN/QL) | DP [V] | CE [eV] (QN/QL) |
|---|---|---|---|---|---|
| Monocrotaline | 3.45 | 326.2 | 120.0/237.1 | 70 | 45/35 |
| Monocrotaline N-oxide | 4.47 | 342.1 | 137.0/120.0 | 135 | 38/44 |
| Jacobine | 4.58 | 352.1 | 155.3/280.4 | 119 | 38/31 |
| Intermedine | 4.64 | 300.2 | 94.2/138.3 | 80 | 33/30 |
| Lycopsamine | 4.78 | 300.2 | 94.2/156.4 | 72 | 34/38 |
| Jacobine N-oxide | 4.85 | 368.2 | 296.2/120.3 | 150 | 34/44 |
| Riddelliine | 4.88 | 350.4 | 120.2/322.2 | 90 | 38/36 |
| Riddelliine N-oxide | 5.03 | 366.1 | 94.1/120.2 | 165 | 72/38 |
| Intermedine N-oxide | 5.16 | 316.2 | 172.0/138.0 | 76 | 38/38 |
| Lycopsamine N-oxide | 5.28 | 316.2 | 172.0/138.0 | 90 | 37/37 |
| Retrorsine | 5.48 | 352.1 | 324.2/138.2 | 126 | 37/39 |
| Retrosine N-oxide | 5.56 | 368.2 | 118.3/120.3 | 105 | 39/41 |
| Seneciphylline | 5.77 | 334.2 | 120.2/306.1 | 100 | 37/36 |
| Heliotrine | 5.87 | 314.2 | 138.0/156.1 | 21 | 29/35 |
| Seneciphylline N-oxide | 5.95 | 350.2 | 120.0/138.0 | 106 | 40/34 |
| Helotrine N-oxide | 6.15 | 330.2 | 172.0/111.0 | 99 | 36/53 |
| Senecionine | 6.40 | 336.2 | 120.0/308.2 | 131 | 40/38 |
| Senecionine N-oxide | 6.53 | 352.2 | 118.0/94.0 | 143 | 43/77 |
| Echimidine | 6.96 | 398.2 | 120.0/220.1 | 77 | 32/26 |
| Senkirkine | 7.15 | 366.2 | 168.1/150.0 | 106 | 40/36 |
| Echimidine N-oxide | 7.48 | 414.2 | 254.1/396.2 | 61 | 40/31 |
| Lasiocarpine | 7.79 | 412.2 | 120.0/336.2 | 30 | 36/27 |
| Lasiocarpine N-oxide | 8.17 | 428.2 | 254.1/410.2 | 100 | 38/32 |
| Petasitenine | 11.30 | 404.3 | 348.1/292.1 | 27 | 17/24 |
| Analyte | LOD (µg/kg) | LOQ (µg/kg) | RSD (Intraday, %) | RSD (Interday, %) | ||||
|---|---|---|---|---|---|---|---|---|
| LOQ | 5 × LOQ | 10 × LOQ | LOQ | 5 × LOQ | 10 × LOQ | |||
| Intermedine | 0.015 | 0.050 | 4.15 | 10.27 | 2.52 | 13.89 | 11.67 | 5.55 |
| Retrorsine | 0.150 | 0.500 | 6.91 | 8.28 | 2.92 | 7.87 | 6.32 | 10.85 |
| Jacobine | 0.150 | 0.500 | 6.09 | 2.69 | 2.20 | 7.15 | 6.30 | 7.21 |
| Lycopsamine | 0.015 | 0.050 | 2.36 | 12.51 | 5.74 | 9.52 | 8.83 | 7.56 |
| Riddelliine | 0.150 | 0.500 | 5.95 | 7.39 | 6.56 | 11.01 | 10.29 | 7.56 |
| Seneciphylline | 0.150 | 0.500 | 5.39 | 7.84 | 2.15 | 9.61 | 9.14 | 8.49 |
| Echimidine | 0.015 | 0.050 | 6.62 | 8.08 | 1.72 | 8.57 | 6.02 | 11.62 |
| Monocrotaline | 0.150 | 0.500 | 3.24 | 3.54 | 2.14 | 4.56 | 9.48 | 7.15 |
| Senecionine | 0.150 | 0.500 | 1.88 | 8.20 | 2.84 | 4.34 | 7.49 | 5.98 |
| Lasiocarpine | 0.015 | 0.050 | 2.12 | 9.73 | 2.20 | 2.80 | 8.28 | 5.35 |
| Heliotrine | 0.015 | 0.050 | 6.27 | 18.23 | 4.13 | 6.92 | 10.86 | 7.26 |
| Senkirkine | 0.015 | 0.050 | 4.53 | 8.92 | 3.07 | 9.45 | 6.39 | 8.31 |
| Petasitenine | 0.300 | 1.000 | 5.14 | 4.85 | 4.82 | 4.87 | 5.39 | 4.44 |
| Lycopsamine N-oxide | 0.075 | 0.250 | 5.05 | 2.54 | 3.33 | 7.67 | 3.98 | 4.64 |
| Intermedine N-oxide | 0.075 | 0.250 | 2.94 | 5.88 | 3.82 | 6.16 | 4.06 | 6.56 |
| Echimidine N-oxide | 0.150 | 0.500 | 3.43 | 5.84 | 0.96 | 4.21 | 8.06 | 3.87 |
| Lasiocarpine N-oxide | 0.075 | 0.250 | 4.99 | 3.40 | 7.99 | 4.65 | 6.39 | 5.39 |
| Monocrotaline N-oxide | 0.150 | 0.500 | 4.44 | 3.51 | 3.79 | 11.36 | 11.05 | 11.96 |
| Senecionine N-oxide | 0.150 | 0.500 | 4.72 | 2.95 | 6.28 | 7.08 | 10.05 | 11.08 |
| Seneciphylline N-oxide | 0.150 | 0.500 | 5.34 | 2.15 | 4.91 | 5.56 | 5.75 | 7.17 |
| Helotrine N-oxide | 0.015 | 0.050 | 7.21 | 5.06 | 2.06 | 6.48 | 9.95 | 11.75 |
| Retrosine N-oxide | 0.075 | 0.250 | 8.09 | 3.97 | 4.33 | 7.05 | 4.31 | 9.35 |
| Jacobine N-oxide | 0.075 | 0.250 | 3.01 | 3.12 | 1.66 | 4.82 | 3.47 | 7.06 |
| Riddelliine N-oxide | 0.150 | 0.500 | 4.69 | 4.86 | 5.92 | 8.22 | 5.69 | 4.59 |
| Sample Type | Sample Number | Type of PA | Concentration (µg/kg) | Concentration of Total PAs (µg/kg) |
|---|---|---|---|---|
| Black tea Green tea Oolong tea | 1 | Echimidine | 0.2 | 0.2 |
| 1 | Seneciphylline | 0.2 | 1.5 | |
| 1 | Seneciphylline | 0.3 | 0.3 | |
| 2 | Lycopsamine N-oxide | 0.9 | 0.9 | |
| 3 | Senkirkine | 1.3 | 1.3 | |
| Amomum kravanh | 1 | Retrosine N-oxide | 92.5 | 92.5 |
| Bitter-bean powder | 1 | Retrorsine, Seneciphylline, Seneciphylline N-oxide, Senecionine, Senecionine N-oxide, Intermedine, Intermedine N-oxide, Lycopsamine | 5.0, 27.4, 48.8, 17.1, 40.9, 1.5, 3.4, 3.6 | 147.8 |
| Angelicae sinensis | 1 | Retrorsine, Retrosine N-oxide | 7.1, 113.2 | 120.3 |
| Codonopsis | 1 | Senecionine N-oxide | 2.4 | 2.4 |
| Senecionis scandentis hebra | 1 | Riddelliine N-oxide, Seneciphylline, Seneciphylline N-oxide, Senecionine, Senecionine N-oxide, Senkirkine, Retrosine N-oxide | 33.7, 362.4, 621.3, 63.4, 83.8, 0.5, 17.2 | 1182.3 |
| Farfarae flos | 1 | Senecionine, Senecionine N-oxide, Retrosine N-oxide | 736.2, 3833.6, 48.8 | 4618.6 |
| Ragwort | 1 | Lycopsamine N-oxide, Senkirkine, Retrosine N-oxide | 50.9, 18.3, 23.8 | 93.0 |
| Arnebiae | 1 | Intermedine, Intermedine N-oxide, Lycopsamine, Lycopsamine N-oxide, Senkirkine, Retrosine N-oxide | 147.8, 712.0, 34.8, 196.0, 1.1, 25.2 | 1117.0 |
| Eupatorii | 1 | Echimidine, Senkirkine, Retrosine N-oxide | 0.4, 1.1, 6.9 | 8.4 |
| Tu-San-Qi | 1 | Seneciphylline, Senecionine, Senecionine N-oxide, Echimidine, Senkirkine, Retrosine N-oxide | 3.1, 4.8, 3.7, 0.4, 16.5, 22.2 | 50.6 |
| Honey | 1 | Retrorsine, Retrosine N-oxide | 3.3, 10.8 | 14.1 |
| 2 | Retrorsine, Retrosine N-oxide | 6.2, 11.0 | 17.2 | |
| 3 | Retrorsine, Intermedine N-oxide, Retrosine N-oxide | 4.8, 0.5, 11.7 | 17.0 | |
| 4 | Retrorsine, Retrosine N-oxide | 4.1, 14.7 | 18.8 | |
| 5 | Retrosine N-oxide | 16.7 | 16.7 | |
| 6 | Retrorsine, Retrosine N-oxide | 3.2, 15.1 | 18.3 | |
| 7 | Retrosine N-oxide | 20.5 | 20.5 | |
| Milk | 1 | Retrorsine, Retrosine N-oxide | 0.8, 23.5 | 24.3 |
| 2 | Retrosine N-oxide | 22.7 | 22.7 | |
| 3 | Echimidine, Retrosine N-oxide | 0.3, 26.2 | 26.5 | |
| 4 | Retrorsine, Retrosine N-oxide | 1.6, 22.2 | 23.9 | |
| 5 | Lycopsamine N-oxide, Retrosine N-oxide | 4.6, 23.9 | 28.6 | |
| 6 | Retrosine N-oxide | 27.9 | 27.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, R.; Peng, J.; Zhu, Y.; Dong, S.; Jiang, X.; Shen, D.; Li, J.; Zhu, P.; Mao, J.; Wang, N.; et al. Quantitative Analysis of Pyrrolizidine Alkaloids in Food Matrices and Plant-Derived Samples Using UHPLC—MS/MS. Foods 2025, 14, 1147. https://doi.org/10.3390/foods14071147
Lin R, Peng J, Zhu Y, Dong S, Jiang X, Shen D, Li J, Zhu P, Mao J, Wang N, et al. Quantitative Analysis of Pyrrolizidine Alkaloids in Food Matrices and Plant-Derived Samples Using UHPLC—MS/MS. Foods. 2025; 14(7):1147. https://doi.org/10.3390/foods14071147
Chicago/Turabian StyleLin, Runfeng, Jing Peng, Yingjie Zhu, Suhe Dong, Xin Jiang, Danning Shen, Jiaxin Li, Peihong Zhu, Jie Mao, Na Wang, and et al. 2025. "Quantitative Analysis of Pyrrolizidine Alkaloids in Food Matrices and Plant-Derived Samples Using UHPLC—MS/MS" Foods 14, no. 7: 1147. https://doi.org/10.3390/foods14071147
APA StyleLin, R., Peng, J., Zhu, Y., Dong, S., Jiang, X., Shen, D., Li, J., Zhu, P., Mao, J., Wang, N., & He, K. (2025). Quantitative Analysis of Pyrrolizidine Alkaloids in Food Matrices and Plant-Derived Samples Using UHPLC—MS/MS. Foods, 14(7), 1147. https://doi.org/10.3390/foods14071147