Determination of Cholesterol Content in Butter by HPLC: Up-to-Date Optimization, and In-House Validation Using Reference Materials


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Standards and Reagents
2.2. Sample Preparation
2.3. Instrument and Chromatographic Conditions
2.4. Calibration Curve
2.5. Method Validation
2.6. Statistical Analysis
3. Results and Discussion
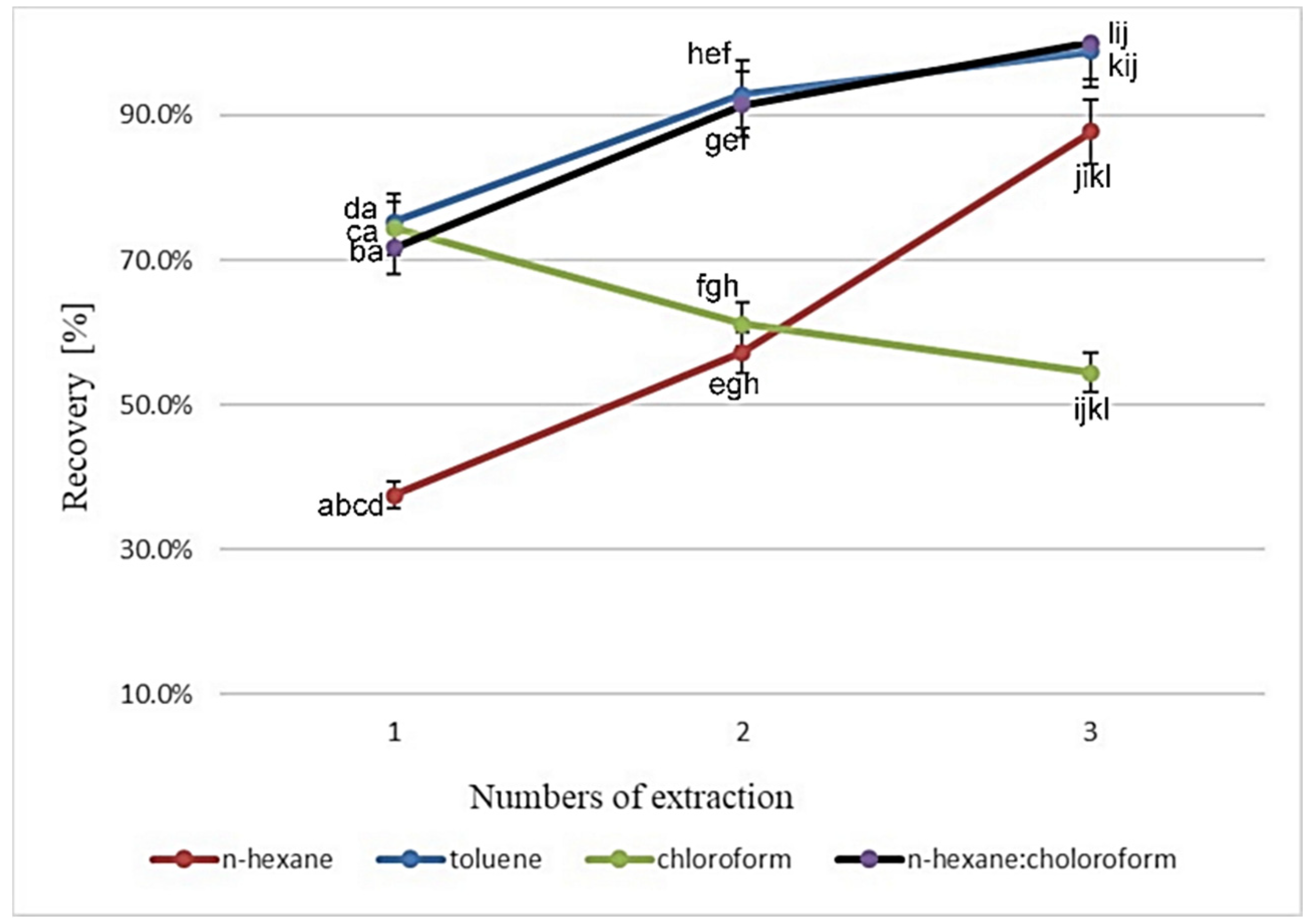
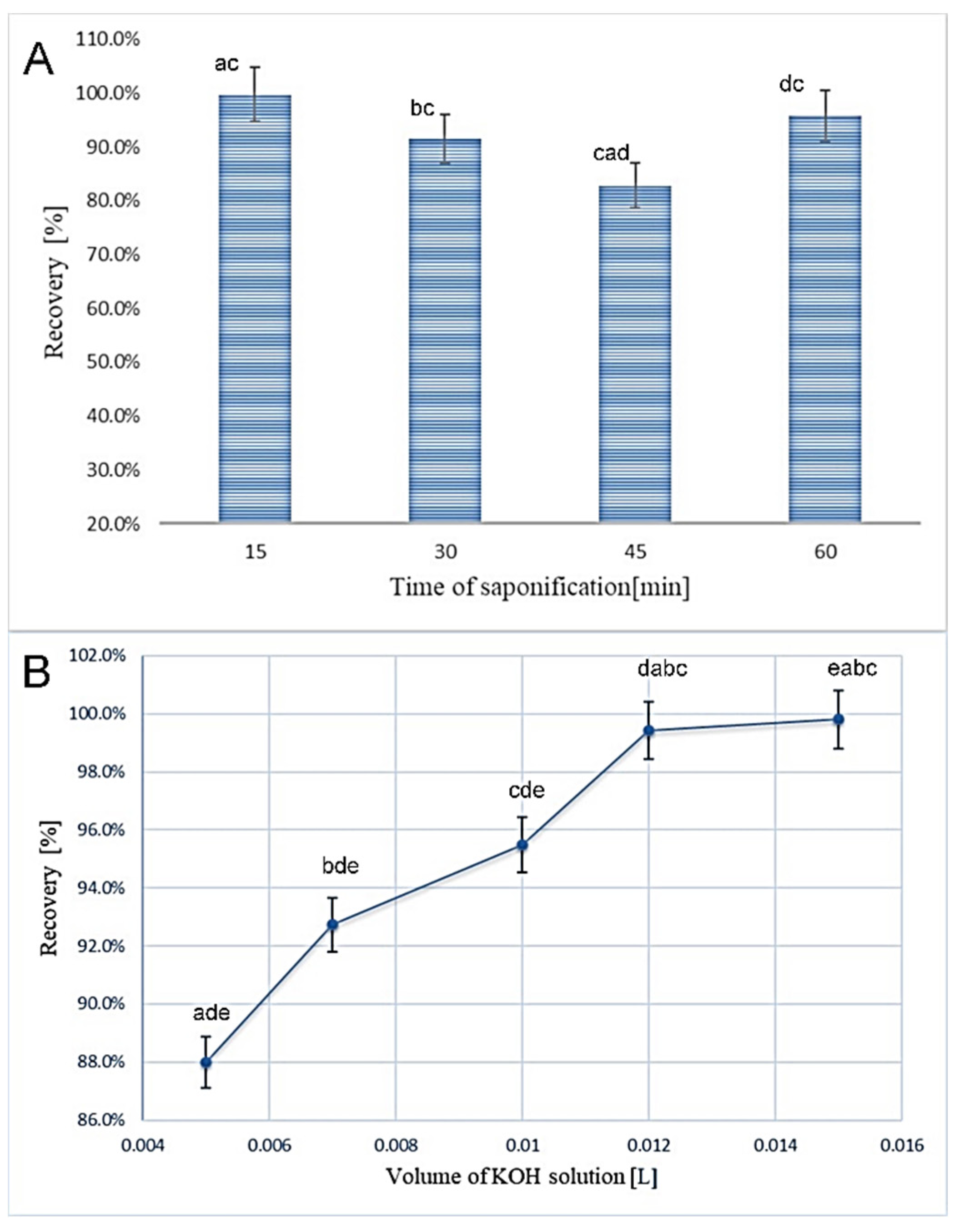
3.1. Optimization of the Sample Preparation
3.2. Method Validation
3.3. Analysis of the Butter
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pereira, P.C. Milk nutritional composition and its role in human health. Nutrition 2014, 30, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Gorassini, A.; Verardo, G.; Fregolent, S.-C.; Bortolomeazzi, R. Rapid determination of cholesterol oxidation products in milk powder based products by reversed phase SPE and HPLC-APCI-MS/MS. Food Chem. 2017, 230, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Shingla, K.M.; Mehta, B.M. Cholesterol and its oxidation products: Occurrence and analysis in milk and milk products. Int. J. Health Anim. Sci. Food Saf. 2018, 5, 13–39. [Google Scholar] [CrossRef]
- Bertolín, J.R.; Joy, M.; Rufino-Moya, P.J.; Lobón, S.; Blanco, M. Simultaneous determination of carotenoids, tocopherols, retinol and cholesterol in ovine lyophilised samples of milk, meat, and liver and in unprocessed/raw samples of fat. Food Chem. 2018, 257, 182–188. [Google Scholar] [CrossRef]
- Dietary Guidelines Advisory Committee. Scientific Report of the 2015 Dietary Guidelines Advisory Committee: Advisory Report to the Secretary of Health and Human Services and the Secretary of Agriculture; U.S. Department of Agriculture, Agricultural Research Service: Washington, DC, USA, 2015. Available online: https://health.gov/our-work/food-nutrition/2015-2020-dietary-guidelines/advisory-report (accessed on 20 July 2020).
- ESC/EAS Guidelines. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Li, L.-H.; Dutkiewicz, E.P.; Huang, Y.-C.; Zhou, H.-B.; Hsu, C.-C. Analytical methods for cholesterol quantification. J. Food Drug Anal. 2019, 27, 375–386. [Google Scholar] [CrossRef]
- Lee, C.-L.; Liao, H.-L.; Lee, W.-C.; Hsu, C.-K.; Hsueh, F.-C.; Pan, J.-Q.; Chu, C.-H.; Wei, C.-T.; Chen, M.-J. Standards and labelling of milk fat and spread products in different countries. J. Food Drug Anal. 2018, 26, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Food and Agriculture Organization of the United Nations. Overview of global dairy market developments in 2018. Dairy Market Review 2019. Available online: http://www.fao.org/publications/card/en/c/CA3879EN/ (accessed on 20 July 2020).
- OECD-FAO Agricultural Outlook 2019–2028; Butter Projections: Consumption, Food (Annex C); Food and Agriculture Organization of the United Nations: Paris, France, 2019; ISBN 9789264312463. [CrossRef]
- Lovegrove, J.A.; Givens, D.I. Dairy food products: Good or bad for cardiometabolic disease? Nutr. Res. Rev. 2016, 29, 249–267. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.H.; Jeong, I.-S.; Kwak, B.M.; Leem, D.; Yoon, T.; Yoon, C.; Jeong, J.; Park, J.M.; Kim, J.M. Rapid determination of cholesterol in milk containing emulsified foods. Food Chem. 2012, 135, 2411–2417. [Google Scholar] [CrossRef]
- Dinh, T.T.N.; Thompson, L.D.; Galyean, M.L.; Brooks, J.C.; Patterson, K.Y.; Boylan, L.M. Cholesterol Content and Methods for Cholesterol Determination in Meat and Poultry. Compr. Rev. Food Sci. Food Saf. 2011, 10, 269–289. [Google Scholar] [CrossRef]
- Borkovcová, I.; Janoušková, E.; Dračková, M.; Janštová, B.; Vorlová, L. Determination of Sterols in Dairy Products and Vegetable Fats by HPLC and GC Methods. Czech. J. Food Sci. 2009, 27, 217–219. [Google Scholar] [CrossRef] [Green Version]
- Bauer, L.C.; de Santana, D.A.; dos Macedo, M.S.; Torres, A.G.; de Souza, N.E.; Simionato, J.I. Method Validation for Simultaneous Determination of Cholesterol and Cholesterol Oxides in Milk by RP-HPLC-DAD. J. Braz. Chem. Soc. 2014, 25, 161–168. [Google Scholar] [CrossRef]
- Albuquerque, T.G.; Oliveira, M.B.P.P.; Sanches-Silva, A.; Costa, H.S. Cholesterol determination in foods: Comparison between high performance and ultra-high performance liquid chromatography. Food Chem. 2016, 193, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Kolarič, L.; Šimko, P. The comparison of HPLC and spectrophotometric method for cholesterol determination. Potravinárstvo Slovak J. Food Sci. 2020, 14, 118–124. [Google Scholar] [CrossRef]
- Oh, H.I.; Shin, T.S.; Chang, E.J. Determination of Cholesterol in Milk and Dairy Products by High-Performance Liquid Chromatography. Asian Australas J. Anim. Sci. 2001, 14, 1465–1469. [Google Scholar] [CrossRef]
- IUPAC Technical Report. Harmonised guidelines for the in-house validation of methods of analysis. In IUPAC/In-House/Budapest; IUPAC: Budapest, Hungary, 2002; Available online: http://old.iupac.org/divisions/V/501/draftoct19.pdf (accessed on 3 July 2020).
- Eurachem. The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics, 2nd ed.; Eurachem: UK, 2014; ISBN 978-91-87461-59-0. Available online: www.eurachem.org (accessed on 29 June 2020).
- Horwitz, W.; Albert, R. The Horwitz Ratio (HorRat): A Useful Index of Method Performance with Respect to Precision. J. AOAC Int. 2006, 89, 1095–1109. [Google Scholar] [CrossRef]
- Dinh, T.T.N.; Blanton, J.R., Jr.; Brooks, J.C.; Miller, M.F.; Thompson, L.D. A simplified method for cholesterol determination in meat and meat products. J. Food Compos. Anal. 2008, 21, 306–314. [Google Scholar] [CrossRef]
- Daneshfar, A.; Khezeli, T.; Lofti, H.J. Determination of cholesterol in food samples using dispersive liquid-liquid microextraction followed by HPLC-UV. J. Chromatogr. B 2009, 877, 456–460. [Google Scholar] [CrossRef]
- Dias, H.M.A.M.; Berbicz, F.; Pedrochi, F.; Baesso, M.L.; Matioli, G. Butter cholesterol removal using different complexation methods with beta-cyclodextrin, and the contribution of photoacoustic spectroscopy to the evaluation of the complex. Food Res. Int. 2010, 43, 1104–1110. [Google Scholar] [CrossRef]
- Ramalho, H.M.M.; Casal, S.; Oliveira, M.B.P.P. Total Cholesterol and Desmosterol Contents in Raw, UHT, Infant Formula Powder and Human Milks Determined by a New Fast Micro-HPLC Method. Food Anal. Methods 2011, 4, 424–430. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Wu, S.-S.; Wu, H.-L. Highly sensitive analysis of cholesterol and sitosterol in foods and human biosamples by liquid chromatography with fluorescence detection. J. Chromatogr. A 2007, 1156, 280–287. [Google Scholar] [CrossRef]
- Adu, J.K.; Amengor, D.K.; Kabiri, N.; Orman, E.; Patamia, S.A.G.; Okrah, B.K. Validation of a Simple and Robust Liebermann-Burchard Colorimetric Method for the Assay of Cholesterol in Selected Milk Products in Ghana. Int. J. Food Sci. 2019, 2019, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Z.; Kao, S.Y.; Jian, H.C.; Yu, Y.M.; Li, J.Y.; Wang, W.H.; Tsai, C.W. Determination of cholesterol and four phytosterols in foods without derivatization by gas chromatography-tandem mass spectrometry. J. Food Drug Anal. 2015, 23, 636–644. [Google Scholar] [CrossRef] [Green Version]
- Shazamawati, Z.H.; Alina, A.R.; Mashitoh, A.S.; Juhana, M.J.T. Cholesterol Oxidation Products Analysis in Meat and Poultry. Middle-East. J. Sci. Res. 2013, 13, 67–78. [Google Scholar] [CrossRef]
- Tahir, M.N.; Kwon, C.; Jeong, D.; Cho, E.; Paik, S.R.; Jung, S. Cholesterol reduction from milk using β-cyclodextrin immobilized on glass. J. Dairy Sci. 2013, 96, 4191–4196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Cervantes, J.; Sánchez-Machado, D.I.; Ríos-Vázquez, N.J. High-performance liquid chromatography method for the simultaneous quantification of retinol, α-tocopherol, and cholesterol in shrimp waste hydrolysate. J. Chromatogr. A 2006, 1105, 135–139. [Google Scholar] [CrossRef]
- Hansen, H.; Wang, T. Does the Saponification-GC Method Underestimate Total Cholesterol Content in Samples Having Considerable Cholesterol Esters? J. Am. Oil Chem. Soc. 2015, 92, 1735–1738. [Google Scholar] [CrossRef]
- Osman, H.; Chin, Y.K. Comparative sensitivities of cholesterol analysis using GC, HPLC and spectrophotometric methods. Malays. J. Anal. Sci. 2006, 10, 205–210. [Google Scholar]
- Ribeiro, T.M.; Brandäo, P.R.G. Development and Validation of Graphitic Carbon Analysis of Graphite Ore Samples. Tecnol. Metal. Mater. Min. 2017, 14, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Commission regulation (EU) No 836/2011 of 19 August 2011 amending Regulation (EC) No 333/2007 laying down the methods of sampling and analysis for the official control of the levels of lead, cadmium, mercury, inorganic tin, 3-MCPD and benzo(a)pyrene in foodstuffs. Off. J. Eur. Union 2011, 215, 9–16. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2011:215:0009:0016:EN:PDF (accessed on 20 July 2020).
- Derewiaka, D.; Sosińska, E.; Obiedziński, M.; Krogulec, A.; Czaplicki, S. Determination of the adulteration of butter. Eur. J. Lipid Sci. Technol. 2011, 113, 1005–1011. [Google Scholar] [CrossRef]
- Gonçalves, M.F.D.; Baggio, S.R. Evaluation of quality of butter from different provenance. Food Sci. Technol. 2012, 32, 629–635. [Google Scholar] [CrossRef] [Green Version]
- Seçkin, A.K.; Gursoy, O.; Kinik, O.; Akbulut, N. Conjugated linoleic acid (CLA) concentration, fatty acid composition and cholesterol of some Turkish dairy products. LWT Food Sci. Technol. 2005, 38, 909–915. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Method linearity | ||||
| Slope a | 11.348 ± 0.026 | |||
| Intercept a | 13.514 ± 3.908 | |||
| Correlation coefficient (r) | 0.999 ± 0.000 | |||
| LOD [mg/kg] | 3.6 | |||
| LOQ [mg/kg] | 11.8 | |||
| Method recoveryb | ||||
| Reference material | Reference value [mg/kg] | Determined value [mg/kg] | Recovery [%] | RSD [%] |
| muva-BU-1312 | 2337 ± 196 | 2395.6 ± 72.6 | 102.5 ± 0.0 | 3.0 |
| muva-BU-1311 | 2334 ± 211 | 2381.6 ± 66.6 | 102.0 ± 0.0 | 2.8 |
| muva-BU-1314 | 2276 ± 135 | 2307.7 ± 58.9 | 101.4 ± 0.0 | 2.6 |
| The amount of cholesterol standard spiked to the butter sample [mg/L] | Determined value [mg/kg] | Recovery [%] | RSD [%] | |
| 0 | 2367.0 ± 29.9 | - | - | |
| 500 | 2460.6 ± 39.0 | 103.9 ± 0.0 | 1.6 | |
| 1000 | 2475.4 ± 31.5 | 104.6 ± 0.0 | 1.3 | |
| 1500 | 2515.9 ± 52.7 | 106.3 ± 0.0 | 2.1 | |
| Method precisionc | ||||
| Reference material | Repeatability | RSD [%] | Intermediate precision [RSD%] | Horrat |
| muva-BU-1312 | Day 1 Day 2 Day 3 | 2.5 2.6 2.2 | 1.8 | 0.01 |
| muva-BU-1311 | Day 1 Day 2 Day 3 | 3.9 2.3 0.6 | 0.9 | 0.01 |
| muva-BU-1314 | Day 1 Day 2 Day 3 | 1.4 3.5 1.9 | 0.8 | 0.01 |
| Dimension of Column(Zorbax Eclipse Plus C18) | Retention Time [min] | Slope | Intercept | LOD [mg/kg] | LOQ [mg/kg] | Cholesterol Content [mg/kg] a |
|---|---|---|---|---|---|---|
| 2.1 × 100 mm, 3.5 µm particle size | 5.6 | 11.348 | 13.514 | 3.6 | 11.8 | 2367.0 ± 29.9 |
| 2.1 × 50 mm, 3.5 µm particle size | 2.2 | 11.349 | 17.320 | 3.0 | 10.0 | 2360.4 ± 27.9 |
| 4.6 × 50 mm, 3.5 µm particle size | 4.4 | 5.713 | 30.071 | 5.5 | 18.4 | 2289.3 ± 53.8 |
| 2.1 × 50 mm, 5.0 µm particle size | 2.2 | 11.480 | 69.452 | 6.8 | 22.5 | 2417.5 ± 92.6 |
| Sample | Cholesterol Content [mg/kg] | RSD [%] |
|---|---|---|
| 1 | 2323.1 ± 29.4 | 1.3 |
| 2 | 2241.1 ± 97.9 | 4.4 |
| 3 | 2582.8 ± 36.5 | 1.4 |
| 4 | 2201.6 ± 52.3 | 2.4 |
| 5 | 1983.7 ± 52.5 | 2.7 |
| 6 | 2252.3 ± 117.5 | 5.2 |
| 7 | 2272.3 ± 87.0 | 3.8 |
| 8 | 2215.9 ± 4.4 | 0.2 |
| 9 | 2460.0 ± 30.3 | 1.2 |
| 10 | 2247.5 ± 119.1 | 5.3 |
| 11 | 2200.8 ± 24.7 | 1.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolarič, L.; Šimko, P. Determination of Cholesterol Content in Butter by HPLC: Up-to-Date Optimization, and In-House Validation Using Reference Materials. Foods 2020, 9, 1378. https://doi.org/10.3390/foods9101378
Kolarič L, Šimko P. Determination of Cholesterol Content in Butter by HPLC: Up-to-Date Optimization, and In-House Validation Using Reference Materials. Foods. 2020; 9(10):1378. https://doi.org/10.3390/foods9101378
Chicago/Turabian StyleKolarič, Lukáš, and Peter Šimko. 2020. "Determination of Cholesterol Content in Butter by HPLC: Up-to-Date Optimization, and In-House Validation Using Reference Materials" Foods 9, no. 10: 1378. https://doi.org/10.3390/foods9101378
APA StyleKolarič, L., & Šimko, P. (2020). Determination of Cholesterol Content in Butter by HPLC: Up-to-Date Optimization, and In-House Validation Using Reference Materials. Foods, 9(10), 1378. https://doi.org/10.3390/foods9101378