Functional Microbiota for Polypeptide Degradation during Hypertonic Moromi-Fermentation of Pixian Broad Bean Paste

Abstract
:1. Introduction
2. Materials and Methods
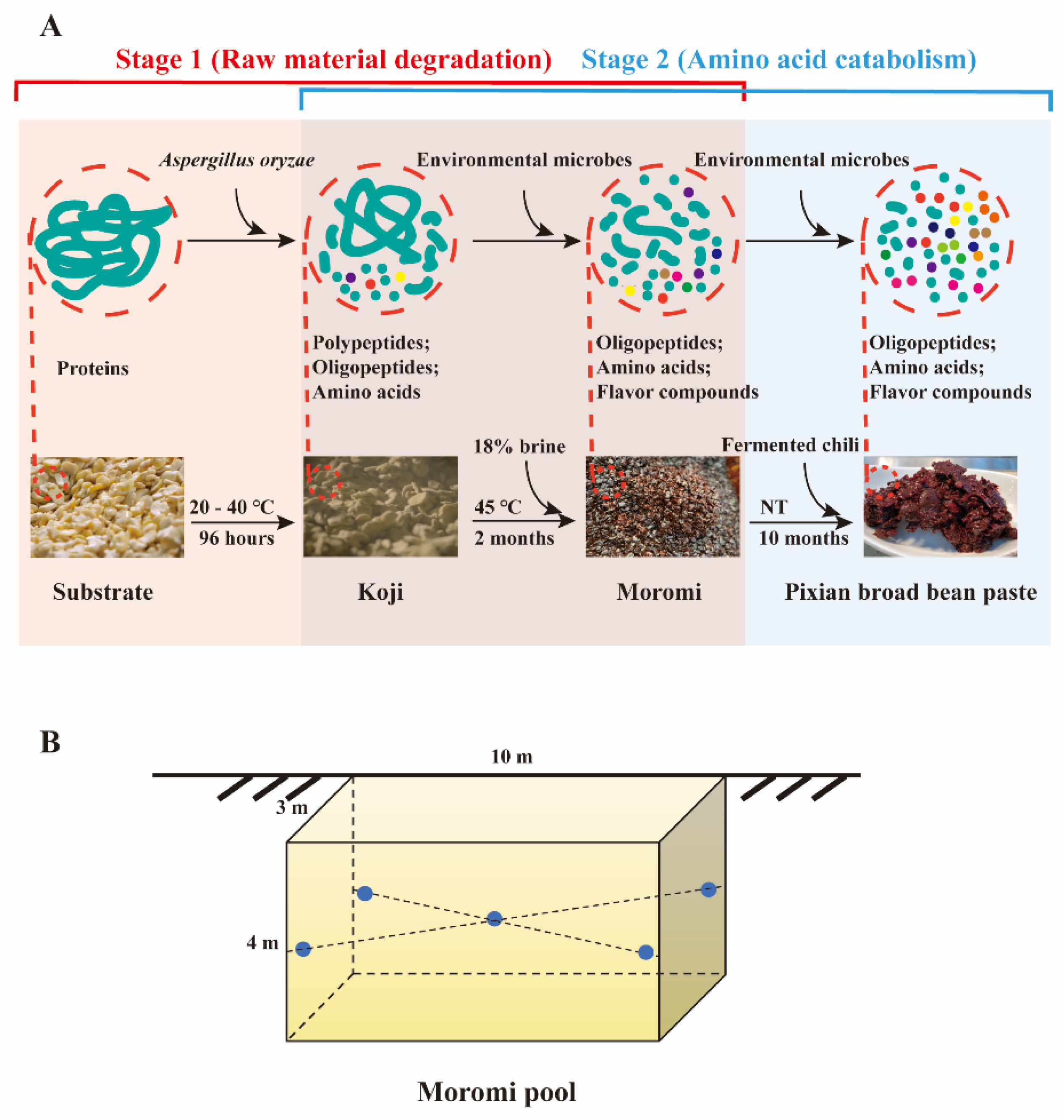
2.1. Sample Collection
2.2. DNA Extraction and Amplification
2.3. High-Throughput Sequencing Analysis
2.4. Quantitative Real-Time PCR (qPCR)
2.5. Organic Acid, Glucose and Ethanol Analysis
2.6. pH, Peptides, Amino Acids and Amino Nitrogen Analysis
2.7. Correlation Analysis among Microbes and Amino Acid Concentration
2.8. Isolation and Characterization of Salt-Tolerant Strains
2.9. Extracellular Proteomic Analysis
2.10. Batch Fermentation to Evaluate the Microbial Function of Polypeptide Degradation
3. Results
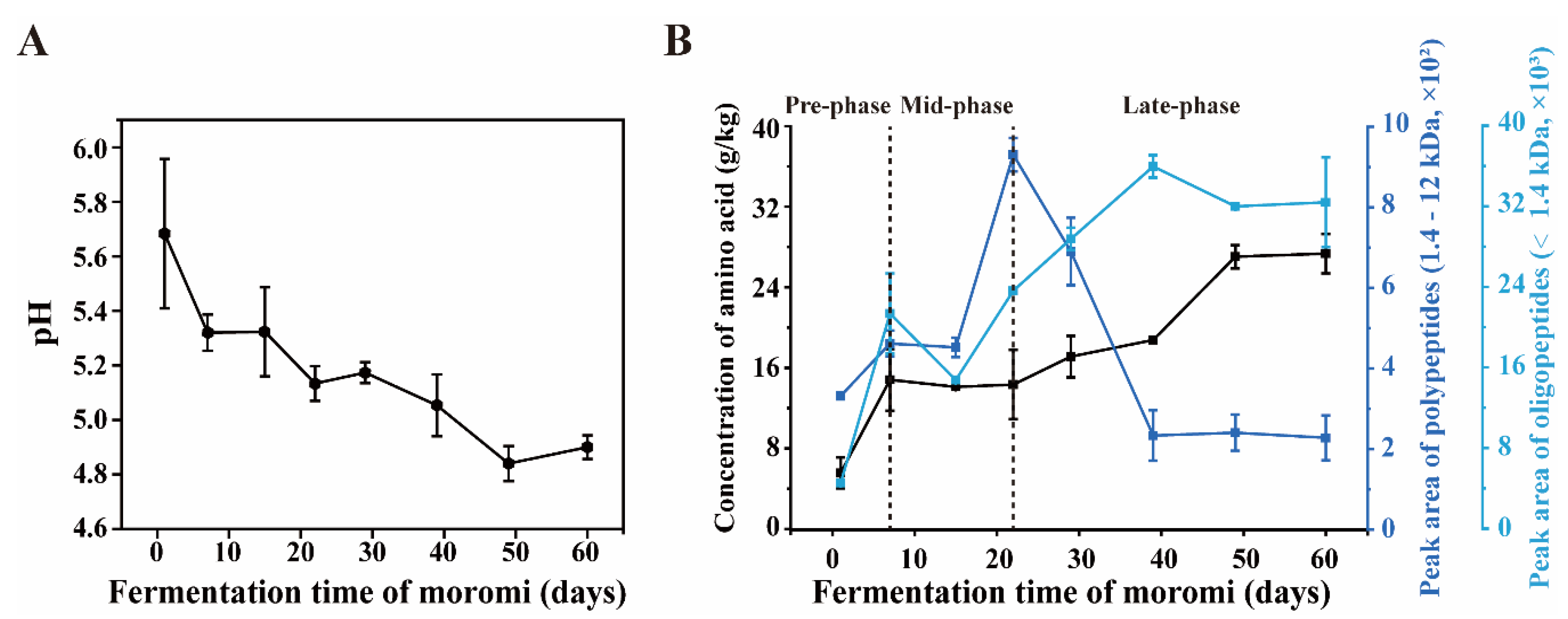
3.1. Amino Acids Accumulate Significantly during Hypertonic Moromi Fermentation
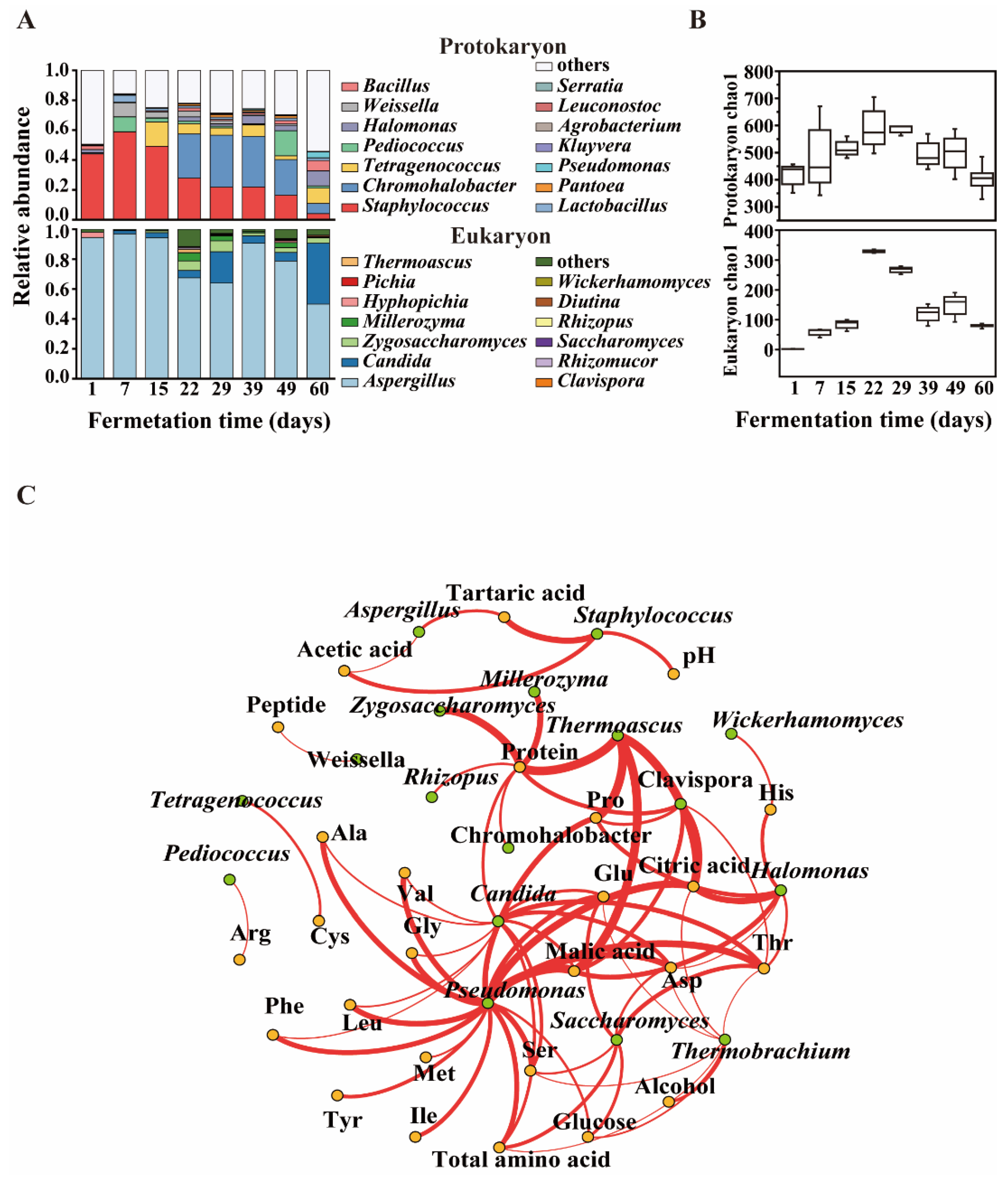
3.2. Microbial Community Structure in Moromi Fermentation
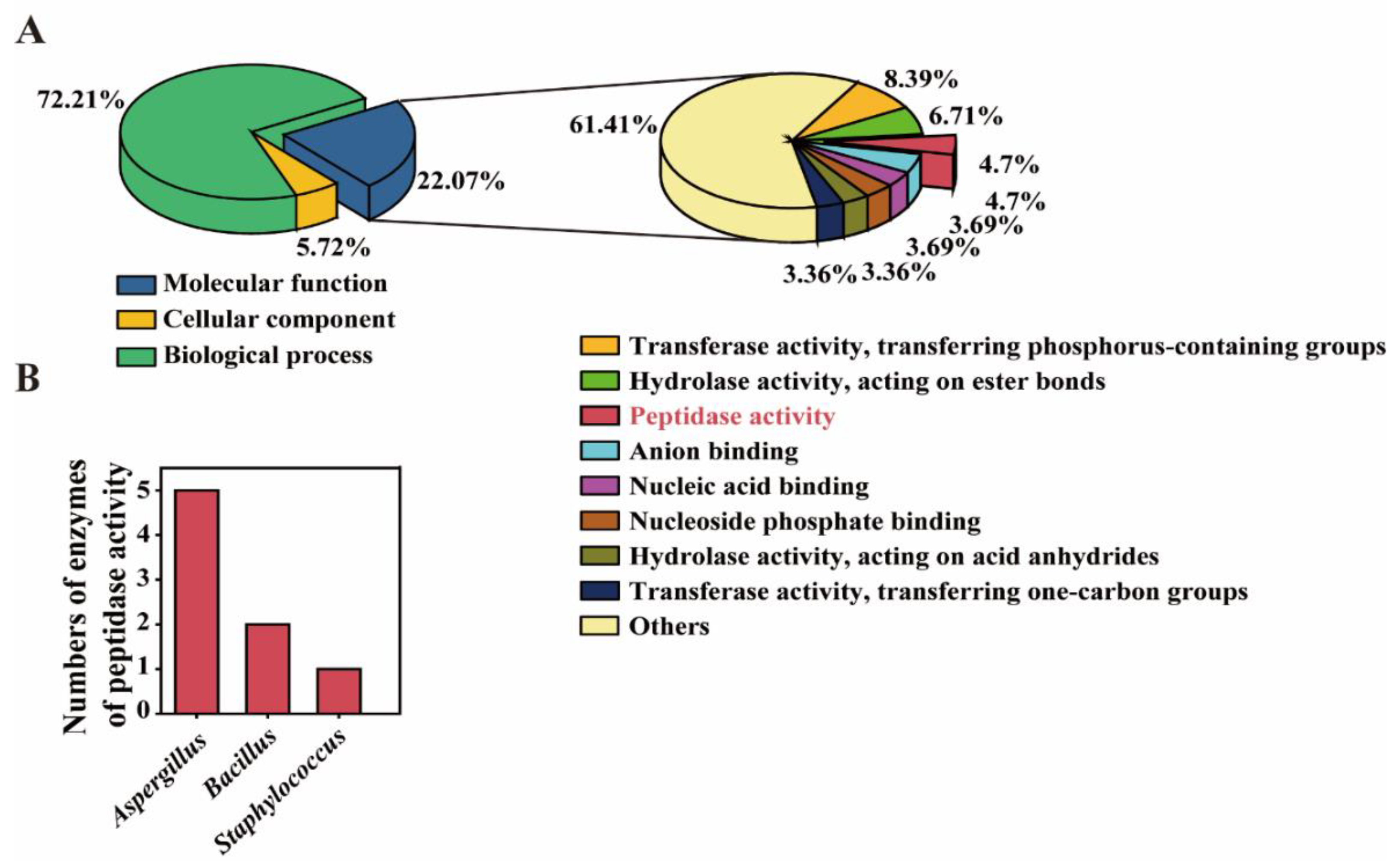
3.3. Potential Functional Microbiota for Polypeptide Degrading Was Found by Using Omics Analysis
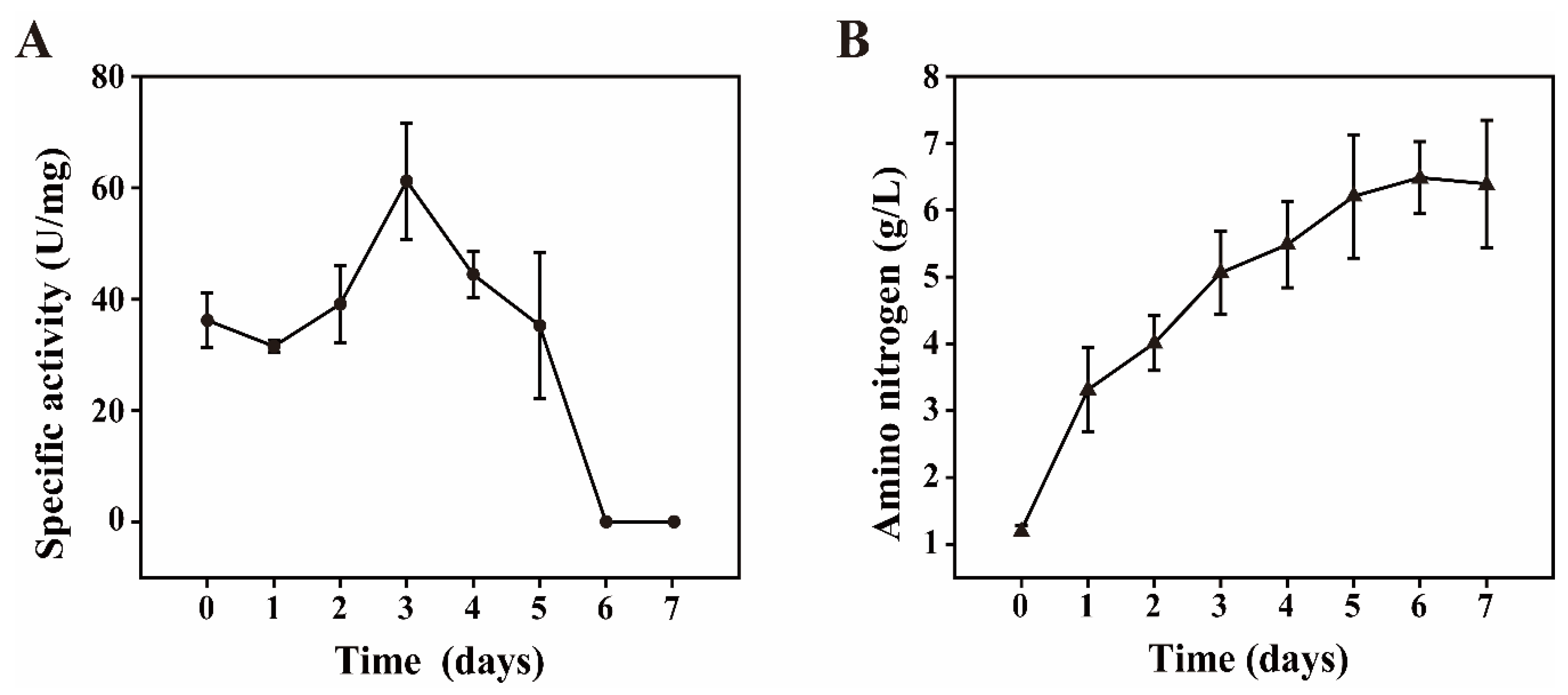
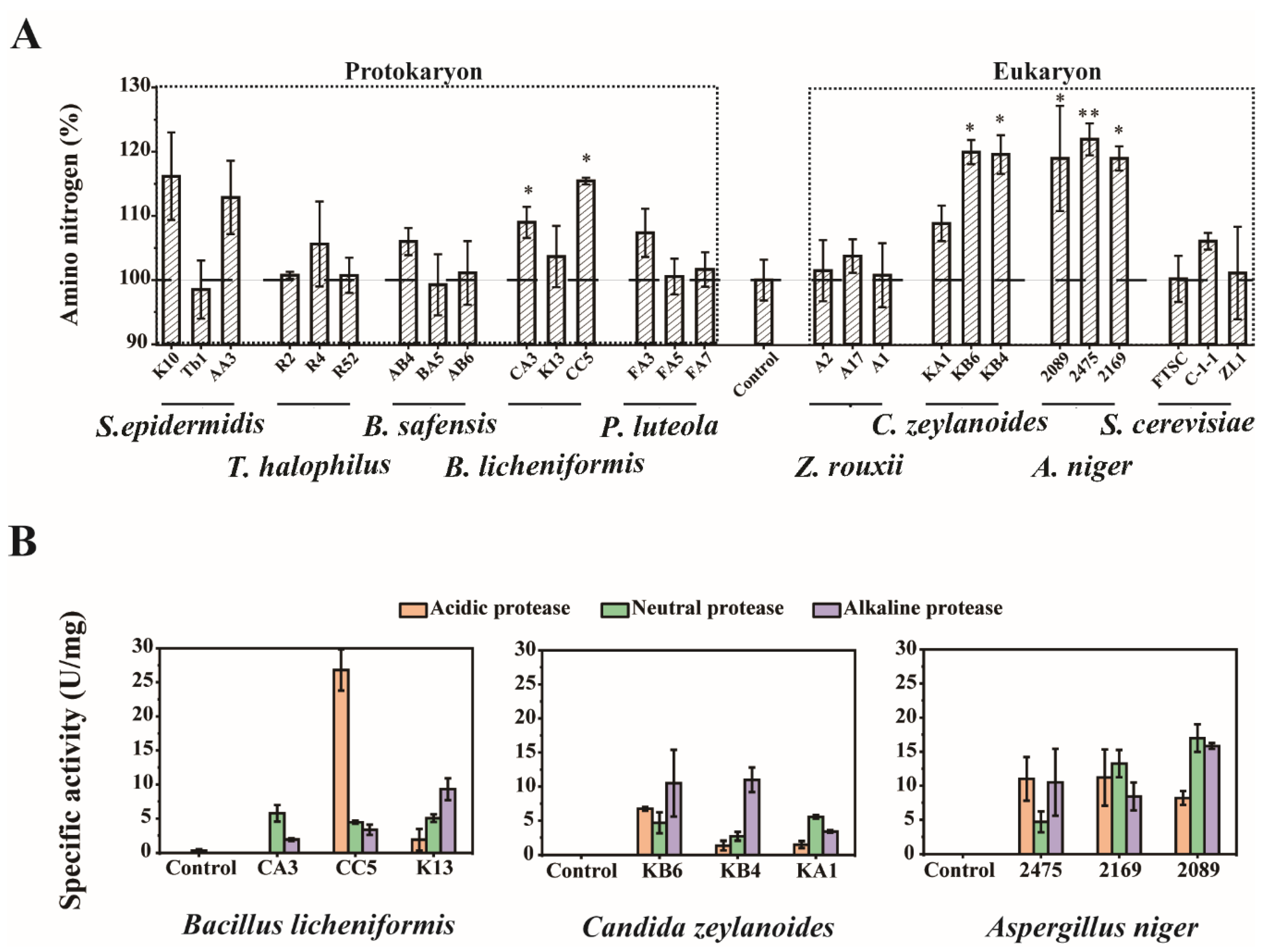
3.4. Batch Fermentation and Enzymatic Analysis Were Used to Determine the Functional Microbiota for Polypeptide Degrading
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ardö, Y. Flavour formation by amino acid catabolism. Biotechnol. Adv. 2006, 24, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.-H.; Green-Johnson, J.; Buckley, N.D.; Lin, Q.-Y. Bioactivity of soy-based fermented foods: A review. Biotechnol. Adv. 2019, 37, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Frérot, E.; Chen, T. Identification and quantitation of new glutamic acid derivatives in soy sauce by UPLC/MS/MS. Chem. Biodivers. 2013, 10, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Park, M.K.; Kim, Y.-S. Comparative metabolic expressions of fermented soybeans according to different microbial starters. Food Chem. 2020, 305, 125461. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, J.; Gan, H.M.; Yin, W.-F.; Chan, K.-G. Microbial succession and the functional potential during the fermentation of Chinese soy sauce brine. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; An, F.; Wu, J.; Liu, Y.; Shi, H.; Wu, R. Meta-omics reveal microbial assortments and key enzymes in bean sauce mash, a traditional fermented soybean product. J. Sci. Food Agric. 2019, 99, 6522–6534. [Google Scholar] [CrossRef]
- Crafack, M.; Keul, H.; Eskildsen, C.E.; Petersen, M.A.; Saerens, S.; Blennow, A.; Skovmand-Larsen, M.; Swiegers, J.H.; Petersen, G.B.; Heimdal, H.; et al. Impact of starter cultures and fermentation techniques on the volatile aroma and sensory profile of chocolate. Food Res. Int. 2014, 63, 306–316. [Google Scholar] [CrossRef]
- Wang, P.; Wu, Q.; Jiang, X.; Wang, Z.; Tang, J.; Xu, Y. Bacillus licheniformis affects the microbial community and metabolic profile in the spontaneous fermentation of Daqu starter for Chinese liquor making. Int. J. Food Microbiol. 2017, 250, 59–67. [Google Scholar] [CrossRef]
- Sanjukta, S.; Rai, A. Production of bioactive peptides during soybean fermentation and their potential health benefits. Trends Food Sci. Technol. 2016, 50, 1–10. [Google Scholar] [CrossRef]
- Lin, H.; Yu, X.; Fang, J.; Lu, Y.; Liu, P.; Xing, Y.; Wang, Q.; Che, Z.; He, Q. Flavor compounds in Pixian broad-bean paste: Non-volatile organic acids and amino acids. Molecules 2018, 23, 1299. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Cao, Y.; Tong, J.; Xu, Y. An alkylpyrazine synthesis mechanism involving l-threonine-3-dehydrogenase describes the production of 2,5-dimethylpyrazine and 2,3,5-trimethylpyrazine by Bacillus subtilis. Appl. Environ. Microbiol. 2019, 85, 01807-19. [Google Scholar] [CrossRef]
- Kitamoto, K. Cell biology of the Koji mold Aspergillus oryzae. Biosci. Biotechnol. Biochem. 2015, 79, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Chi, Y.; Lv, Y.; Yang, G.; He, Q. Evolution of the volatile flavor compounds of Chinese horse bean-chili-paste. LWT 2019, 102, 131–135. [Google Scholar] [CrossRef]
- Lu, Y.; Tan, X.; Lv, Y.; Yang, G.; Chi, Y.; He, Q. Physicochemical properties and microbial community dynamics during Chinese horse bean-chili-paste fermentation, revealed by culture-dependent and culture-independent approaches. Food Microbiol. 2020, 85, 103309. [Google Scholar] [CrossRef]
- Cui, C.; Zhao, M.; Li, D.; Zhao, H.; Sun, W. Biochemical changes of traditional Chinese-type soy sauce produced in four seasons during processing. CyTA J. Food 2013, 12, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.S.; Lee, Y.B.; Lee, D.S.; Chung, S.K. Quality evaluation of Korean soy sauce fermented in Korean earthenware (Onggi) with different glazes. Int. J. Food Sci. Technol. 2006, 41, 1158–1163. [Google Scholar] [CrossRef]
- Ma, Y.-L.; Wang, J.-H.; Cheng, Y.-Q.; Yin, L.-J.; Li, L.-T. Some biochemical and physical changes during manufacturing of grey sufu, a traditional Chinese fermented soybean curd. Int. J. Food Eng. 2013, 9, 45–54. [Google Scholar] [CrossRef]
- Udomsil, N.; Rodtong, S.; Choi, Y.J.; Hua, Y.; Yongsawatdigul, J. Use of Tetragenococcus halophilus as a starter culture for flavor improvement in fish sauce fermentation. J. Agric. Food Chem. 2011, 59, 8401–8408. [Google Scholar] [CrossRef]
- Liu, M.; Nauta, A.; Francke, C.; Siezen, R.J. Comparative genomics of enzymes in flavor-forming pathways from amino acids in lactic acid bacteria. Appl. Environ. Microbiol. 2008, 74, 4590–4600. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Chen, C.; Lei, Z. Meta-omics insights in the microbial community profiling and functional characterization of fermented foods. Trends Food Sci. Technol. 2017, 65, 23–31. [Google Scholar] [CrossRef]
- Wang, X.; Du, H.; Zhang, Y.; Xu, Y. Environmental microbiota drives microbial succession and metabolic profiles during Chinese liquor fermentation. Appl. Environ. Microbiol. 2017, 84, e02369-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Tian, X.; Ma, L.; Feng, B.; Liu, Q.; Yuan, L.; Fan, C.; Huang, H.; Yang, Q.; Xiaoling, Z.; et al. Biodiversity of the symbiotic bacteria associated with toxic marine dinoflagellate Alexandrium tamarense. J. Biosci. Med. 2015, 3, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Toju, H.; Tanabe, A.S.; Yamamoto, S.; Sato, H. High-coverage ITS primers for the DNA-based identification of Ascomycetes and Basidiomycetes in environmental samples. PLoS ONE 2012, 7, e40863. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wu, Q.; Nie, Y.; Wu, J.; Xu, Y. Construction of synthetic microbiota for reproducible flavor compound metabolism in Chinese light-aroma-type liquor produced by solid-state fermentation. Appl. Environ. Microbiol. 2019, 85, e03090-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; I Gordon, J.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N.; Jackson, J.A.; Vilgalys, R.; Jackson, R.B. Assessment of soil microbial community structure by use of taxon-specific quantitative PCR assays. Appl. Environ. Microbiol. 2005, 71, 4117–4120. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wu, Q.; Wang, P.; Lin, J.; Huang, L.; Xu, Y. Synergistic effect in core microbiota associated with sulfur metabolism in spontaneous Chinese liquor fermentation. Appl. Environ. Microbiol. 2017, 83, e01475-17. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Feng, S.; Wu, Q.; Huang, H.; Chen, Z.; Li, S.; Xu, Y. Raw material regulates flavor formation via driving microbiota in Chinese liquor fermentation. Front. Microbiol. 2019, 10, 1520. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Chen, L.; Xu, Y. Yeast community associated with the solid state fermentation of traditional Chinese Maotai-flavor liquor. Int. J. Food Microbiol. 2013, 166, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Mao, X.; Zhang, J.; Du, G.; Chen, J. Effective biodegradation of chicken feather waste by co-cultivation of keratinase producing strains. Microb. Cell Fact. 2019, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Vidyalakshmi, R.; Paranthaman, R.; Sureshkumar, K. Simultaneous determination of 17 amino acids in microbial pigments from Monascus spp by UHPLC amino acid analyzer using pre-column derivatization. Int. J. Curr. Microbiol. Appl. Sci. 2016, 5, 354–362. [Google Scholar] [CrossRef]
- Liu, F.; Chen, Z.; Shao, J.; Wang, C.; Zhan, C. Effect of fermentation on the peptide content, phenolics and antioxidant activity of defatted wheat germ. Food Biosci. 2017, 20, 141–148. [Google Scholar] [CrossRef]
- Su, G.; Ren, J.; Zhao, M.; Sun, D.-W. Comparison of superdex peptide HR 10/30 column and TSK Gel G2000 SWXL column for molecular weight distribution analysis of protein hydrolysates. Food Bioprocess Technol. 2012, 6, 3620–3626. [Google Scholar] [CrossRef]
- Cui, R.-Y.; Zheng, J.; Wu, C.; Zhou, R.-Q. Effect of different halophilic microbial fermentation patterns on the volatile compound profiles and sensory properties of soy sauce moromi. Eur. Food Res. Technol. 2014, 239, 321–331. [Google Scholar] [CrossRef]
- Wang, B.; Wu, Q.; Xu, Y.; Sun, B. Specific volumetric weight-driven shift in microbiota compositions with saccharifying activity change in starter for Chinese Baijiu fermentation. Front. Microbiol. 2018, 9, 2349. [Google Scholar] [CrossRef]
- Ma, J.; Chen, T.; Wu, S.; Yang, C.; Bai, M.; Shu, K.; Li, K.; Zhang, G.; Jin, Z.; He, F.; et al. iProX: An integrated proteome resource. Nucleic Acids Res. 2018, 47, D1211–D1217. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Cui, C.; Zhao, H.; Gao, X.; Zhao, M.; Sun, W. Effect of koji fermentation on generation of volatile compounds in soy sauce production. Int. J. Food Sci. Technol. 2012, 48, 609–619. [Google Scholar] [CrossRef]
- Wang, D.; Zheng, Z.; Feng, J.; Zhan, X.-B.; Zhang, L.M.; Wu, J.R.; Lin, C.-C. A high salt tolerant neutral protease from Aspergillus oryzae: Purification, characterization and kinetic properties. Appl. Biochem. Microbiol. 2013, 49. [Google Scholar] [CrossRef]
- Li, Z.; Rui, J.; Li, X.; Li, J.; Dong, L.; Huang, Q.; Huang, C.; Wang, Z.; Li, L.; Xuan, P.; et al. Bacterial community succession and metabolite changes during doubanjiang-meju fermentation, a Chinese traditional fermented broad bean (Vicia faba L.) paste. Food Chem. 2017, 218, 534–542. [Google Scholar] [CrossRef]
- Xie, M.; Wu, J.; An, F.; Yue, X.; Tao, D.; Wu, R.; Lee, Y.-K. An integrated metagenomic/metaproteomic investigation of microbiota in dajiang-meju, a traditional fermented soybean product in Northeast China. Food Res. Int. 2019, 115, 414–424. [Google Scholar] [CrossRef]
- Kwak, M.-J.; Kong, H.G.; Choi, K.; Kwon, S.-K.; Song, J.Y.; Lee, J.; Lee, P.A.; Choi, S.Y.; Seo, M.; Lee, H.J.; et al. Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 2018, 36, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zheng, X.; Ma, X.; Jiang, R.; Zhou, W.; Zhou, S.; Zhang, Y.; Lei, S.; Wang, S.; Kuang, J.; et al. Theabrownin from Pu-erh tea attenuates hypercholesterolemia via modulation of gut microbiota and bile acid metabolism. Nat. Commun. 2019, 10, 4971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Zheng, Z.; Feng, J.; Zhan, X.-B.; Zhang, L.; Wu, J.-R.; Lin, C.-C.; Zhu, L. Influence of sodium chloride on thermal denaturation of a high-salt-tolerant neutral protease from Aspergillus oryzae. Food Sci. Biotechnol. 2013, 22, 1–7. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, C.; Li, S.; Wang, X.; Yao, Y. Exploring the flavor formation mechanism under osmotic conditions during soy sauce fermentation in Aspergillus oryzae by proteomic analysis. Food Funct. 2020, 11, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liang, X.; Zhang, H.; Feng, W.; Liu, Y.; Zhang, F.; Linhardt, R.J. A comparative secretome analysis of industrial Aspergillus oryzae and its spontaneous mutant ZJGS-LZ-21. Int. J. Food Microbiol. 2017, 248, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tramper, J. Koji–where East meets West in fermentation. Biotechnol. Adv. 2013, 31, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Guo, G.; Wang, C.; Lin, Y.; Wang, X.; Zhao, M.; Guo, Y.; He, M.; Zhang, Y.; Pan, L. Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res. 2010, 38, 5075–5087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bautista-Gallego, J.; Rantsiou, K.; Garrido-Fernández, A.; Cocolin, L.; Arroyo-López, F.N. Salt reduction in vegetable fermentation: Feality or desire? J. Food Sci. 2013, 78, R1095–R1100. [Google Scholar] [CrossRef]
- Li, Z.; Dong, L.; Huang, Q.; Wang, X. Bacterial communities and volatile compounds in Doubanjiang, a Chinese traditional red pepper paste. J. Appl. Microbiol. 2016, 120, 1585–1594. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolated Strains | Accession Number | LBM ID | Description |
|---|---|---|---|
| Staphylococcus epidermidis K10 | MN428237.1 | LBM 19032 | |
| S. epidermidis AA3 | CP045648.1 | LBM 19054 | Isolated from Pixian broad bean paste |
| S. epidermidis Tb1 | KM253004.1 | LBM 19116 | |
| Tetragenococcus halophilus R2 | MK063722.1 | LBM 19170 | |
| T. halophilus R4 | MK063724.1 | LBM 19172 | LBM strain collection |
| T. halophilus R52 | GQ150506.1 | LBM 19211 | (isolated from fermented food) |
| Bacillus safensis AB4 | MN704448.1 | LBM 19056 | |
| B. safensis AB6 | MN704552.1 | LBM 19057 | Isolated from Pixian broad bean paste |
| B. safensis AA5 | MN704552.1 | LBM 19058 | |
| Bacillus licheniformis CA3 | KU922147.1 | LBM 19067 | |
| B. licheniformis CC5 | CP045814.1 | LBM 19115 | Isolated from Pixian broad bean paste |
| B. licheniformis K13 | MK414971.1 | LBM 19103 | |
| Pseudomonas luteola FA3 | CP044086.1 | LBM 19128 | |
| P. luteola FA7 | NR_114215.1 | LBM 19222 | Isolated from Pixian broad bean paste |
| P. luteola FA9 | CP044085.1 | LBM 19223 | |
| Zygosaccharomyces rouxii A2 | AB363049.1 | LBM 29111 | |
| Z. rouxii A17 | MH669504.1 | LBM 29104 | LBM strain collection |
| (isolated from fermented food) | |||
| Z. rouxii A1 | MK294302.1 | LBM 29101 | |
| Candida zelanoides KA1 | KX376266.1 | LBM 29033 | |
| C. zelanoides KB6 | MN073455.1 | LBM 29038 | Isolated from Pixian broad bean paste |
| C. zelanoides KB4 | MK613257.1 | LBM 29037 | |
| Aspergillus niger 2089 | MN420840.1 | LBM 39001 | |
| A. niger 2475 | MN535797.1 | LBM 39003 | LBM strain collection |
| (isolated from fermented food) | |||
| A. niger 2169 | MN611114.1 | LBM 39002 | |
| Saccharomyces cerevisiae FTSC | MN585900.1 | LBM 27012 | |
| S. cerevisiae C-1-1 | MN585905.1 | LBM 27046 | LBM strain collection |
| (isolated from fermented food) | |||
| S. cerevisiae ZL1 | MN158119.1 | LBM 27145 | |
| Aspergillus oryzae HN 3.042 | CGMCC 3.951 |
| Amino Acid | Fermentation Time (Day) | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 7 | 15 | 22 | 29 | 39 | 49 | 60 | |
| Threonine | 0.20 ± 0.02 | 0.68 ± 0.16 | 0.48 ± 0.23 | 0.71 ± 0.19 | 0.84 ± 0.10 | 1.17 ± 0.30 | 1.30 ± 0.08 | 1.33 ± 0.12 |
| Serine | 0.01 ± 0.01 | 0.07 ± 0.01 | 0.09 ± 0.04 | 0.07 ± 0.02 | 0.15 ± 0.03 | 0.21 ± 0.05 | 0.25 ± 0.09 | 0.29 ± 0.14 |
| Glycine | 0.14 ± 0.03 | 0.35 ± 0.08 | 0.56 ± 0.27 | 0.42 ± 0.11 | 0.51 ± 0.07 | 0.72 ± 0.22 | 0.83 ± 0.07 | 0.89 ± 0.09 |
| Alanine | 0.29 ± 0.04 | 0.81 ± 0.16 | 1.17 ± 0.48 | 0.92 ± 0.22 | 1.06 ± 0.13 | 1.52 ± 0.40 | 1.70 ± 0.11 | 1.85 ± 0.11 |
| Proline | 0.55 ± 0.02 | 0.50 ± 0.14 | 0.89 ± 0.43 | 0.91 ± 0.42 | 0.81 ± 0.20 | 1.11 ± 0.30 | 1.36 ± 0.10 | 1.29 ± 0.11 |
| Glutamic acid | 0.81 ± 0.13 | 2.64 ± 0.54 | 2.43 ± 0.05 | 2.47 ± 0.51 | 2.95 ± 0.39 | 3.18 ± 0.08 | 4.45 ± 0.15 | 4.50 ± 0.21 |
| Aspartic acid | 0.46 ± 0.02 | 1.15 ± 0.18 | 1.26 ± 0.03 | 1.21 ± 0.30 | 1.76 ± 0.26 | 2.46 ± 0.66 | 2.81 ± 0.16 | 2.91 ± 0.22 |
| Lysine | 0.55 ± 0.17 | 1.80 ± 0.46 | 2.31 ± 1.19 | 1.43 ± 0.33 | 1.53 ± 0.23 | 2.03 ± 0.69 | 2.26 ± 0.04 | 2.33 ± 0.12 |
| Histidine | 0.14 ± 0.04 | 0.05 ± 0.01 | 0.06 ± 0.03 | 0.05 ± 0.01 | 0.06 ± 0.01 | 0.53 ± 0.64 | 0.35 ± 0.35 | 1.01 ± 0.07 |
| Arginine | 1.18 ± 0.40 | 2.47 ± 0.48 | 2.70 ± 1.06 | 1.73 ± 0.24 | 1.73 ± 0.26 | 2.13 ± 0.40 | 2.66 ± 0.10 | 2.62 ± 0.06 |
| Methionine | 0.09 ± 0.02 | 0.20 ± 0.04 | 0.30 ± 0.14 | 0.20 ± 0.05 | 0.25 ± 0.04 | 0.33 ± 0.11 | 0.33 ± 0.04 | 0.30 ± 0.05 |
| Isoleucine | 0.23 ± 0.08 | 0.56 ± 0.14 | 0.91 ± 0.42 | 0.68 ± 0.21 | 0.80 ± 0.11 | 1.12 ± 0.34 | 1.30 ± 0.13 | 1.29 ± 0.15 |
| Leucine | 0.38 ± 2.09 | 1.06 ± 0.23 | 1.66 ± 0.70 | 1.23 ± 0.34 | 1.38 ± 0.17 | 1.92 ± 0.60 | 2.29 ± 0.20 | 2.30 ± 0.28 |
| Tyrosine | 0.23 ± 0.12 | 0.75 ± 0.13 | 0.92 ± 0.27 | 0.72 ± 0.15 | 0.85 ± 0.09 | 1.16 ± 0.33 | 1.38 ± 0.09 | 1.40 ± 0.09 |
| Phenylalanine | 0.29 ± 0.09 | 0.66 ± 0.15 | 1.02 ± 0.41 | 0.75 ± 0.22 | 0.81 ± 0.09 | 1.09 ± 0.33 | 1.37 ± 0.15 | 1.37 ± 0.17 |
| Valine | 0.33 ± 0.11 | 0.79 ± 0.17 | 1.18 ± 0.51 | 0.88 ± 0.23 | 0.98 ± 0.13 | 1.39 ± 0.42 | 1.58 ± 0.14 | 1.63 ± 0.20 |
| Cysteine | 0.00 ± 0.00 | 0.01 ± 0.01 | 0.02 ± 0.02 | 0.01 ± 0.01 | 0.01 ± 0.01 | 0.02 ± 0.02 | 0.01 ± 0.01 | 0.01 ± 0.01 |
| Total | 5.41 ± 1.34 | 14.55 ± 3.09 | 17.96 ± 6.27 | 14.39 ± 3.55 | 16.49 ± 2.32 | 22.09 ± 5.89 | 26.24 ± 2.02 | 27.32 ± 2.18 |
| Enzyme | Accession Number | Abundance % | Description | Source |
|---|---|---|---|---|
| Neutral protease2 | sp|B8NWE1|NPIIB_ASPFN | 6.96 × 10−4 | EC:3.4.24.39. metalloendopeptidase activity | Aspergillus flavus |
| Subtilisin-like serine protease | XP_022386201.1 | 1.50 × 10−5 | Serine-type endopeptidase activity | Aspergillus bombycis |
| Carboxypeptidase | XP_022388195.1 | 1.09 × 10−5 | Serine-type carboxypeptidase activity | A. bombycis |
| Aspartic proteinase | CEL07294.1 | 4.32 × 10−5 | Aspartic-type endopeptidase activity | Aspergillus calidoustus |
| Aspergillopepsin-1 | sp|B8NLY9|PEPA_ASPFN | 1.07 × 10−3 | EC:3.4.23.18. aspartic-type endopeptidase activity | A. flavus |
| Cyanophycinase | WP_099000026.1 | 2.04 × 10−5 | Serine-type peptidase activity | Bacillus aryabhattai |
| CocE/NonD family hydrolase | WP_066152222.1 | 2.29 × 10−6 | Dipeptidyl-peptidase activity | Bacillus krulwichiae |
| Methionyl aminopeptidase | WP_103328270.1 | 2.25 × 10−4 | EC:3.4.11.18. metalloaminopeptidase activity | Staphylococcus petrasii |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Bao, Y.; Chen, H.; Huang, J.; Xu, Y. Functional Microbiota for Polypeptide Degradation during Hypertonic Moromi-Fermentation of Pixian Broad Bean Paste. Foods 2020, 9, 930. https://doi.org/10.3390/foods9070930
Zhang L, Bao Y, Chen H, Huang J, Xu Y. Functional Microbiota for Polypeptide Degradation during Hypertonic Moromi-Fermentation of Pixian Broad Bean Paste. Foods. 2020; 9(7):930. https://doi.org/10.3390/foods9070930
Chicago/Turabian StyleZhang, Lijie, Yida Bao, Haifeng Chen, Jiaquan Huang, and Yan Xu. 2020. "Functional Microbiota for Polypeptide Degradation during Hypertonic Moromi-Fermentation of Pixian Broad Bean Paste" Foods 9, no. 7: 930. https://doi.org/10.3390/foods9070930
APA StyleZhang, L., Bao, Y., Chen, H., Huang, J., & Xu, Y. (2020). Functional Microbiota for Polypeptide Degradation during Hypertonic Moromi-Fermentation of Pixian Broad Bean Paste. Foods, 9(7), 930. https://doi.org/10.3390/foods9070930