
A Fully Automated Online SPE-LC-MS/MS Method for the Determination of 10 Pharmaceuticals in Wastewater Samples

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Safety
2.3. Instrumentation
2.4. Sample Collection and Preparation
2.5. Optimization of LC-MS Conditions
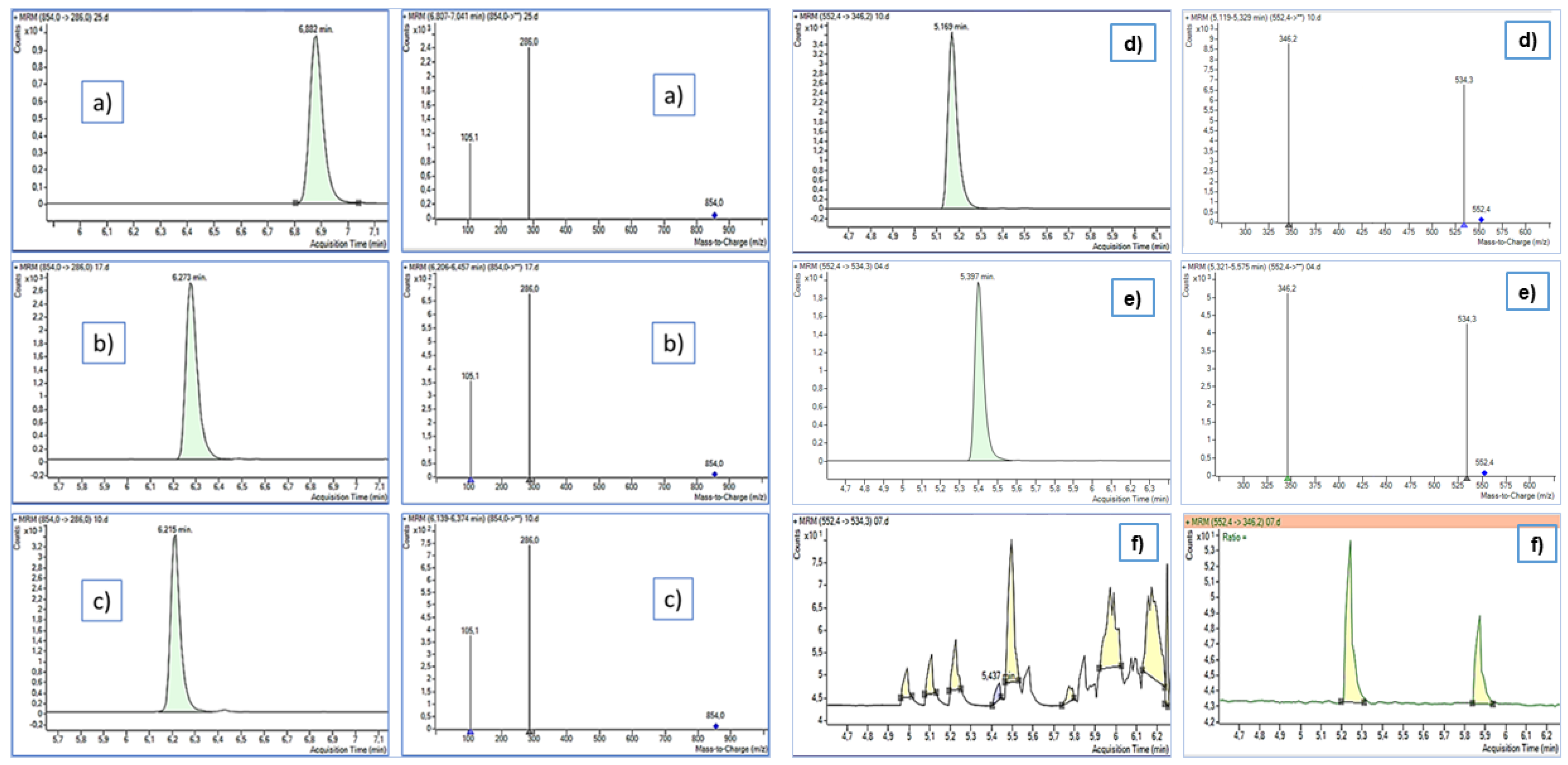
2.5.1. Optimization of the MRM Method
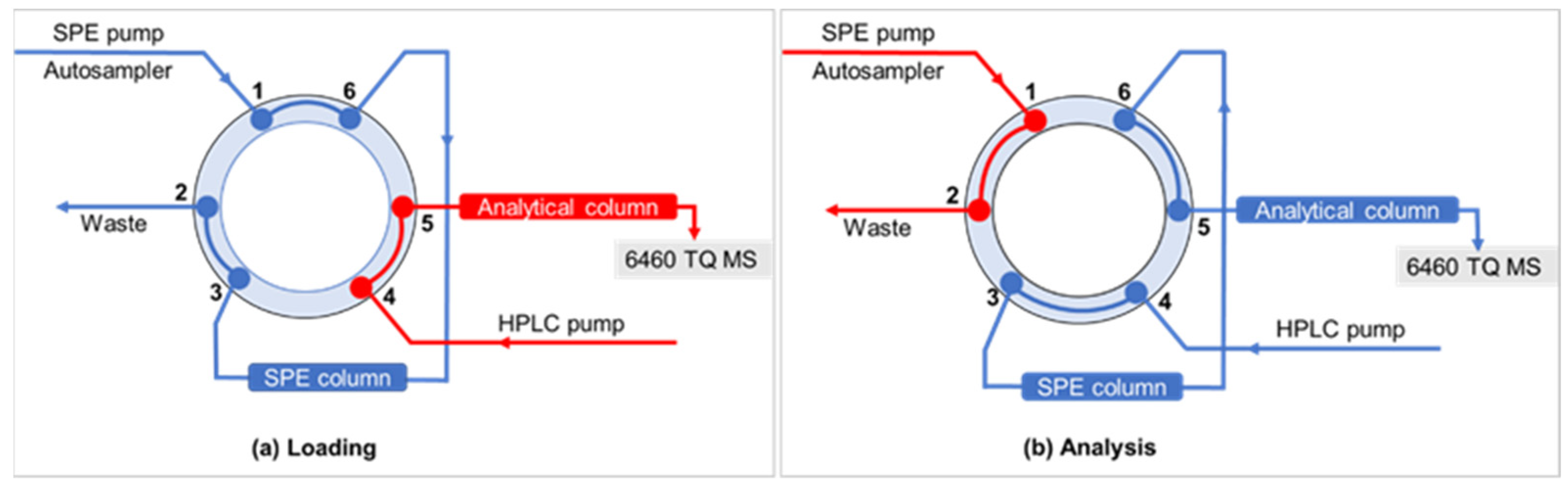
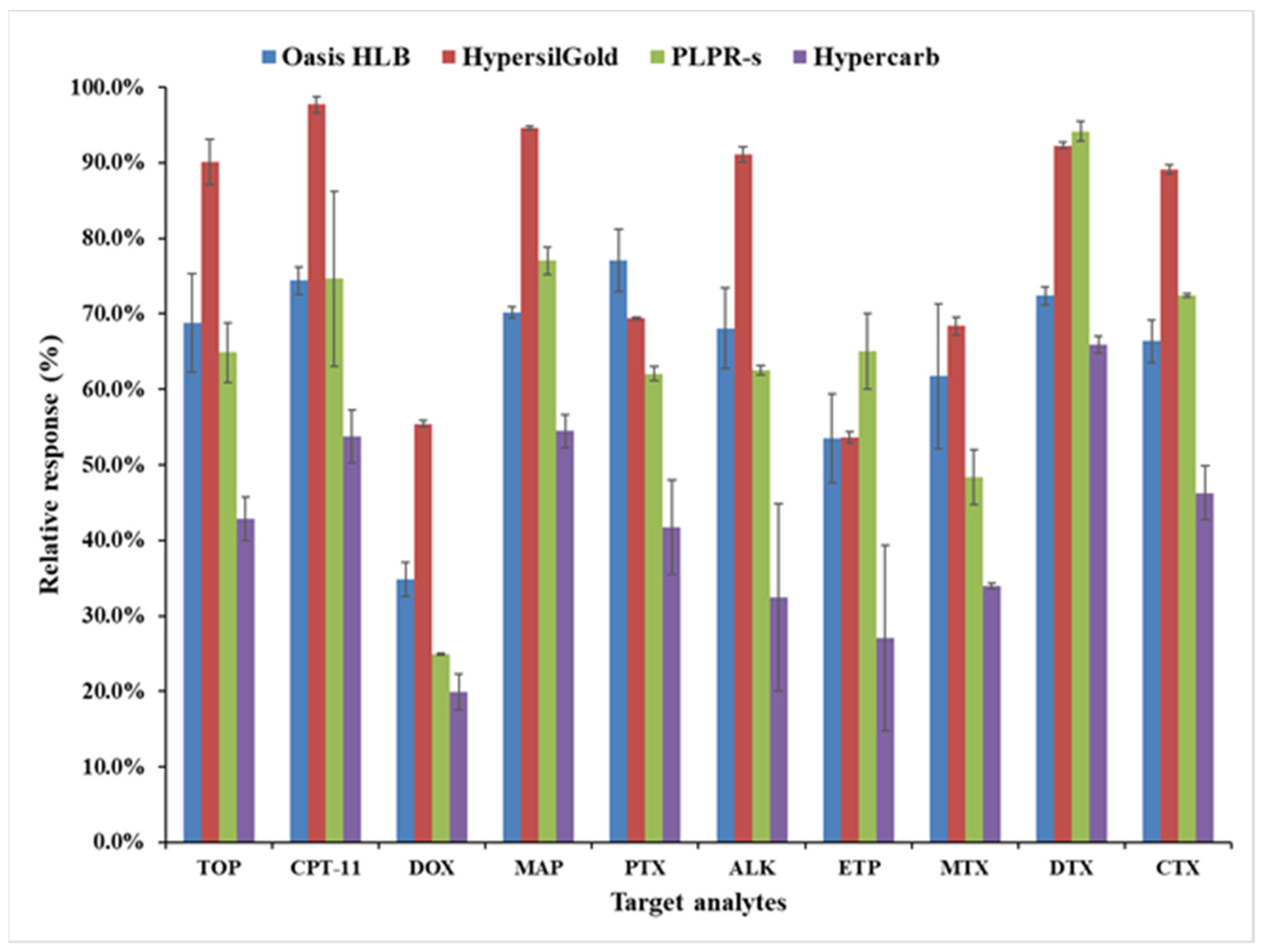
2.5.2. Selection of Online SPE Cartridges
2.5.3. Optimization of LC-Dependent Conditions
2.5.4. Method Validation
3. Results and Discussion
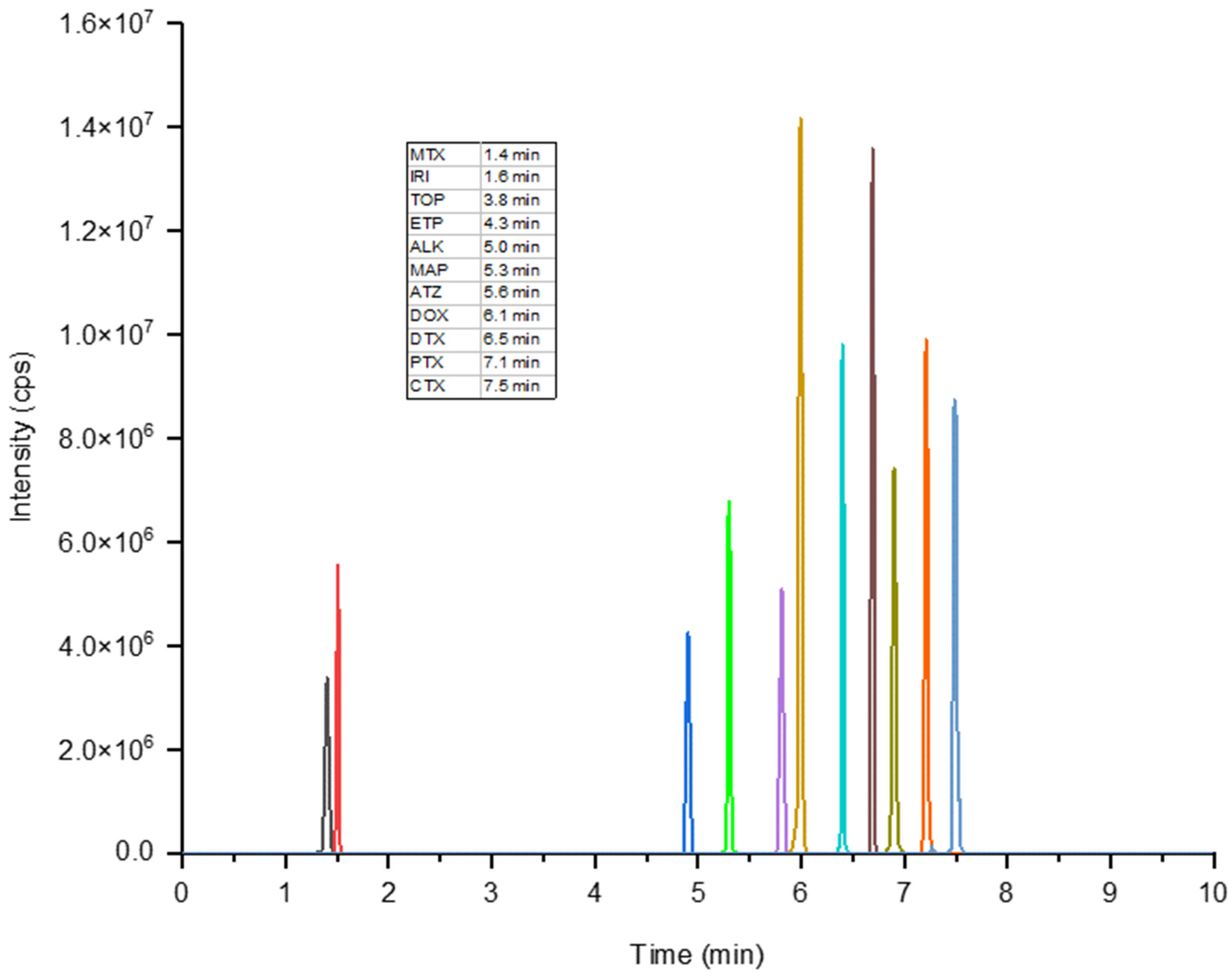
3.1. Optimization of the HPLC-MS/MS
3.2. Optimization of the Online SPE
3.3. Method Validation
3.4. Analysis of Real Water Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rozman, D.; Hrkal, Z.; Váňa, M.; Vymazal, J.; Boukalová, Z. Occurrence of Pharmaceuticals in Wastewater and Their Interaction with Shallow Aquifers: A Case Study of Horní Beřkovice, Czech Republic. Water 2017, 9, 218. [Google Scholar] [CrossRef]
- Shraim, A.; Diab, A.; Alsuhaimi, A.; Niazy, E.; Metwally, M.; Amad, M.; Sioud, S.; Dawoud, A. Analysis of some pharmaceuticals in municipal wastewater of Almadinah Almunawarah. Arab. J. Chem. 2017, 10, S719–S729. [Google Scholar] [CrossRef] [Green Version]
- Ferrando-Climent, L.; Rodriguez-Mozaz, S.; Barceló, D. Development of a UPLC-MS/MS method for the determination of ten anticancer drugs in hospital and urban wastewaters, and its application for the screening of human metabolites assisted by information-dependent acquisition tool (IDA) in sewage samples. Anal. Bioanal. Chem. 2013, 405, 5937–5952. [Google Scholar] [CrossRef] [Green Version]
- Patrolecco, L.; Capri, S.; Ademollo, N. Occurrence of selected pharmaceuticals in the principal sewage treatment plants in Rome (Italy) and in the receiving surface waters. Environ. Sci. Pollut. Res. 2014, 22, 5864–5876. [Google Scholar] [CrossRef]
- Papagiannaki, D.; Morgillo, S.; Bocina, G.; Calza, P.; Binetti, R. Occurrence and Human Health Risk Assessment of Pharmaceuticals and Hormones in Drinking Water Sources in the Metropolitan Area of Turin in Italy. Toxics 2021, 9, 88. [Google Scholar] [CrossRef]
- De Jongh, C.M.; Kooij, P.J.; de Voogt, P.; ter Laak, T.L. Screening and human health risk assessment of pharmaceuticals and their transformation products in Dutch surface waters and drinking water. Sci. Total Environ. 2012, 427, 70–77. [Google Scholar] [CrossRef]
- Grabicova, K.; Lindberg, R.H.; Östman, M.; Grabic, R.; Randak, T.; Larsson, D.J.; Fick, J. Tissue-specific bioconcentration of antidepressants in fish exposed to effluent from a municipal sewage treatment plant. Sci. Total Environ. 2014, 488–489, 46–50. [Google Scholar] [CrossRef]
- Jureczko, M.; Kalka, J. Cytostatic pharmaceuticals as water contaminants. Eur. J. Pharmacol. 2020, 866, 172816. [Google Scholar] [CrossRef]
- Deo, R.P.; Halden, R.U. Pharmaceuticals in the Built and Natural Water Environment of the United States. Water 2013, 5, 1346–1365. [Google Scholar] [CrossRef]
- Petrie, B.; Barden, R.; Kasprzyk-Hordern, B. A review on emerging contaminants in wastewaters and the environment: Current knowledge, understudied areas and recommendations for future monitoring. Water Res. 2015, 72, 3–27. [Google Scholar] [CrossRef]
- Papageorgiou, M.; Kosma, C.; Lambropoulou, D. Seasonal occurrence, removal, mass loading and environmental risk assessment of 55 pharmaceuticals and personal care products in a municipal wastewater treatment plant in Central Greece. Sci. Total Environ. 2016, 543, 547–569. [Google Scholar] [CrossRef]
- Vymazal, J.; Březinová, T.D.; Koželuh, M.; Kule, L. Occurrence and removal of pharmaceuticals in four full-scale constructed wetlands in the Czech Republic–the first year of monitoring. Ecol. Eng. 2017, 98, 354–364. [Google Scholar] [CrossRef]
- Chander, V.; Sharma, B.; Negi, V.; Aswal, R.S.; Singh, P.; Singh, R.; Dobhal, R. Pharmaceutical Compounds in Drinking Water. J. Xenobiot. 2016, 6, 5774. [Google Scholar] [CrossRef] [PubMed]
- Heath, E.; Česen, M.; Negreira, N.; De Alda, M.L.; Climent, L.F.; Bláhová, L.; Nguyen, T.V.; Adahchour, M.; Ruebel, A.; Llewellyn, N.; et al. First inter-laboratory comparison exercise for the determination of anticancer drugs in aqueous samples. Environ. Sci. Pollut. Res. 2015, 23, 14692–14704. [Google Scholar] [CrossRef]
- Carioli, G.; Bertuccio, P.; Boffetta, P.; Levi, F.; La Vecchia, C.; Negri, E.; Malvezzi, M. European cancer mortality predictions for the year 2020 with a focus on prostate cancer. Ann. Oncol. 2020, 31, 650–658. [Google Scholar] [CrossRef]
- Kondor, A.C.; Molnár, É.; Vancsik, A.; Filep, T.; Szeberényi, J.; Szabó, L.; Maász, G.; Pirger, Z.; Weiperth, A.; Ferincz, Á.; et al. Occurrence and health risk assessment of pharmaceutically active compounds in riverbank filtrated drinking water. J. Water Process Eng. 2021, 41, 102039. [Google Scholar] [CrossRef]
- Besse, J.-P.; Latour, J.-F.; Garric, J. Anticancer drugs in surface waters: What can we say about the occurrence and environmental significance of cytotoxic, cytostatic and endocrine therapy drugs? Environ. Int. 2012, 39, 73–86. [Google Scholar] [CrossRef]
- Booker, V.; Halsall, C.; Llewellyn, N.; Johnson, A.; Williams, R. Prioritising anticancer drugs for environmental monitoring and risk assessment purposes. Sci. Total Environ. 2014, 473–474, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Nassour, C.; Barton, S.J.; Nabhani-Gebara, S.; Saab, Y.; Barker, J. Occurrence of anticancer drugs in the aquatic environment: A systematic review. Environ. Sci. Pollut. Res. 2020, 27, 1339–1347. [Google Scholar] [CrossRef] [Green Version]
- Santos, L.H.M.L.M.; Araújo, A.N.; Fachini, A.; Pena, A.; Delerue-Matos, C.; Montenegro, M.C.B.S.M. Ecotoxicological aspects related to the presence of pharmaceuticals in the aquatic environment. J. Hazard. Mater. 2010, 175, 45–95. [Google Scholar] [CrossRef] [Green Version]
- Lutterbeck, C.A.; Baginska, E.; Machado, E.L.; Kümmerer, K. Removal of the anti-cancer drug methotrexate from water by advanced oxidation processes: Aerobic biodegradation and toxicity studies after treatment. Chemosphere 2015, 141, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Kümmerer, K.; Al-Ahmad, A.; Mersch-Sundermann, V. Biodegradability of some antibiotics, elimination of the genotoxicity and affection of wastewater bacteria in a simple test. Chemosphere 2000, 40, 701–710. [Google Scholar] [CrossRef]
- Kovalova, L.; McArdell, C.S.; Hollender, J. Challenge of high polarity and low concentrations in analysis of cytostatics and metabolites in wastewater by hydrophilic interaction chromatography/tandem mass spectrometry. J. Chromatogr. A 2009, 1216, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Shao, B.; Zhang, J.; Li, K. A Preliminary Study on the Occurrence of Cytostatic Drugs in Hospital Effluents in Beijing, China. Bull. Environ. Contam. Toxicol. 2010, 84, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Martín, J.; Camacho-Muñoz, D.; Santos, J.L.; Aparicio, I.; Alonso, E. Simultaneous determination of a selected group of cytostatic drugs in water using high-performance liquid chromatography–triple-quadrupole mass spectrometry. J. Sep. Sci. 2011, 34, 3166–3177. [Google Scholar] [CrossRef]
- Rabii, F.W.; Segura, P.A.; Fayad, P.B.; Sauvé, S. Determination of six chemotherapeutic agents in municipal wastewater using online solid-phase extraction coupled to liquid chromatography-tandem mass spectrometry. Sci. Total Environ. 2014, 487, 792–800. [Google Scholar] [CrossRef]
- Negreira, N.; de Alda, M.L.; Barceló, D. Cytostatic drugs and metabolites in municipal and hospital wastewaters in Spain: Filtration, occurrence, and environmental risk. Sci. Total Environ. 2014, 497–498, 68–77. [Google Scholar] [CrossRef]
- Buerge, I.J.; Buser, H.-R.; Poiger, T.; Müller, M.D. Occurrence and Fate of the Cytostatic Drugs Cyclophosphamide and Ifosfamide in Wastewater and Surface Waters. Environ. Sci. Technol. 2006, 40, 7242–7250. [Google Scholar] [CrossRef]
- Valcárcel, Y.; Alonso, S.G.; Rodríguez-Gil, J.; Gil, A.; Catalá, M. Detection of pharmaceutically active compounds in the rivers and tap water of the Madrid Region (Spain) and potential ecotoxicological risk. Chemosphere 2011, 84, 1336–1348. [Google Scholar] [CrossRef]
- Negreira, N.; de Alda, M.L.; Barceló, D. On-line solid phase extraction–liquid chromatography–tandem mass spectrometry for the determination of 17 cytostatics and metabolites in waste, surface and ground water samples. J. Chromatogr. A 2013, 1280, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Franquet-Griell, H.; Gómez-Canela, C.; Ventura, F.; Lacorte, S. Anticancer drugs: Consumption trends in Spain, prediction of environmental concentrations and potential risks. Environ. Pollut. 2017, 229, 505–515. [Google Scholar] [CrossRef]
- Yin, J.; Yang, Y.; Li, K.; Zhang, J.; Shao, B. Analysis of anticancer drugs in sewage water by selective SPE and UPLC-ESI-MS-MS. J. Chromatogr. Sci. 2010, 48, 781–789. [Google Scholar] [CrossRef] [Green Version]
- Waldmeier, F.; Glaenzel, U.; Wirz, B.; Oberer, L.; Schmid, D.; Seiberling, M.; Valencia, J.; Riviere, G.-J.; End, P.; Vaidyanathan, S. Absorption, Distribution, Metabolism, and Elimination of the Direct Renin Inhibitor Aliskiren in Healthy Volunteers. Drug Metab. Dispos. 2007, 35, 1418–1428. [Google Scholar] [CrossRef] [Green Version]
- Isidori, M.; Lavorgna, M.; Russo, C.; Kundi, M.; Žegura, B.; Novak, M.; Filipič, M.; Mišík, M.; Knasmueller, S.; de Alda, M.L.; et al. Chemical and toxicological characterisation of anticancer drugs in hospital and municipal wastewaters from Slovenia and Spain. Environ. Pollut. 2016, 219, 275–287. [Google Scholar] [CrossRef] [Green Version]
- Vieno, N.; Hallgren, P.; Wallberg, P.; Pyhälä, M.; Zandaryaa, S.; Commission, B.M.E.P. Pharmaceuticals in the Aquatic Environment of the Baltic Sea Region: A Status Report; UNESCO Publishing: Paris, France, 2017; Volume 1. [Google Scholar]
- Fáberová, M.; Bodik, I.; Ivanová, L.; Grabic, R.; Mackuľak, T. Frequency and use of pharmaceuticals in selected Slovakian town via wastewater analysis. Mon. Chem. Chem. Mon. 2017, 148, 441–448. [Google Scholar] [CrossRef]
- Singer, H.; Wössner, A.E.; McArdell, C.S.; Fenner, K. Rapid Screening for Exposure to “Non-Target” Pharmaceuticals from Wastewater Effluents by Combining HRMS-Based Suspect Screening and Exposure Modeling. Environ. Sci. Technol. 2016, 50, 6698–6707. [Google Scholar] [CrossRef]
- Gosetti, F.; Belay, M.H.; Marengo, E.; Robotti, E. Development and validation of a UHPLC-MS/MS method for the identification of irinotecan photodegradation products in water samples. Environ. Pollut. 2020, 256, 113370. [Google Scholar] [CrossRef]
- Gonçalves, N.P.; Varga, Z.; Bouchonnet, S.; Dulio, V.; Alygizakis, N.; Bello, F.D.; Medana, C.; Calza, P. Study of the photoinduced transformations of maprotiline in river water using liquid chromatography high-resolution mass spectrometry. Sci. Total Environ. 2021, 755, 143556. [Google Scholar] [CrossRef]
- Calza, P.; Medana, C.; Padovano, E.; Giancotti, V.; Minero, C. Fate of selected pharmaceuticals in river waters. Environ. Sci. Pollut. Res. 2012, 20, 2262–2270. [Google Scholar] [CrossRef]
- Gonçalves, N.P.; Iezzi, L.; Belay, M.H.; Dulio, V.; Alygizakis, N.; Bello, F.D.; Medana, C.; Calza, P. Elucidation of the photoinduced transformations of Aliskiren in river water using liquid chromatography high-resolution mass spectrometry. Sci. Total Environ. 2021, 800, 149547. [Google Scholar] [CrossRef]
- Vazquez-Roig, P.; Andreu, V.; Onghena, M.; Blasco, C.; Picó, Y. Assessment of the occurrence and distribution of pharmaceuticals in a Mediterranean wetland (L’Albufera, Valencia, Spain) by LC-MS/MS. Anal. Bioanal. Chem. 2011, 400, 1287–1301. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Zhang, C.; Wang, Y.; Guo, Y.; Huang, P.; Zhao, L. Simultaneous determination of 12 pharmaceuticals in water samples by ultrasound-assisted dispersive liquid-liquid microextraction coupled with ultra-high performance liquid chromatography with tandem mass spectrometry. Anal. Bioanal. Chem. 2016, 408, 8099–8109. [Google Scholar] [CrossRef]
- Suazo, F.; Vásquez, J.; Retamal, M.; Ascar, L.; Giordano, A. Pharmaceutical compounds determination in water samples: Comparison between solid phase extraction and STIR Bar sorptive extraction. J. Chil. Chem. Soc. 2017, 62, 3597–3601. [Google Scholar] [CrossRef] [Green Version]
- Kachhawaha, A.S.; Nagarnaik, P.M.; Jadhav, M.; Pudale, A.; Labhasetwar, P.K.; Banerjee, K. Optimization of a Modified QuEChERS Method for Multiresidue Analysis of Pharmaceuticals and Personal Care Products in Sewage and Surface Water by LC-MS/MS. J. AOAC Int. 2017, 100, 592–597. [Google Scholar] [CrossRef]
- Ohoro, C.; Adeniji, A.; Okoh, A. Distribution and Chemical Analysis of Pharmaceuticals and Personal Care Products (PPCPs) in the Environmental Systems: A Review. Int. J. Environ. Res. Public Health 2019, 16, 3026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, K.; Park, M.; Huang, Z.; Jia, A.; Flores, G.; Lee, H.; Snyder, S. A review of extraction methods for the analysis of pharmaceuticals in environmental waters. Crit. Rev. Environ. Sci. Technol. 2020, 50, 2271–2299. [Google Scholar] [CrossRef]
- Sadutto, D.; Picó, Y. Sample Preparation to Determine Pharmaceutical and Personal Care Products in an All-Water Matrix: Solid Phase Extraction. Molecules 2020, 25, 5204. [Google Scholar] [CrossRef] [PubMed]
- Togola, A.; Baran, N.; Coureau, C. Advantages of online SPE coupled with UPLC/MS/MS for determining the fate of pesticides and pharmaceutical compounds. Anal. Bioanal. Chem. 2013, 406, 1181–1191. [Google Scholar] [CrossRef] [Green Version]
- Papagiannaki, D.; Belay, M.H.; Gonçalves, N.P.; Robotti, E.; Bianco-Prevot, A.; Binetti, R.; Calza, P. From monitoring to treatment, how to improve water quality: The pharmaceuticals case. Chem. Eng. J. Adv. 2022, 10, 100245. [Google Scholar] [CrossRef]
- Patrolecco, L.; Ademollo, N.; Grenni, P.; Tolomei, A.; Caracciolo, A.B.; Capri, S. Simultaneous determination of human pharmaceuticals in water samples by solid phase extraction and HPLC with UV-fluorescence detection. Microchem. J. 2013, 107, 165–171. [Google Scholar] [CrossRef]
- Bozyiğit, G.D.; Ayyıldız, M.F.; Chormey, D.S.; Engin, G.O.; Bakırdere, S. Trace level determination of eleven nervous system–active pharmaceutical ingredients by switchable solvent-based liquid-phase microextraction and gas chromatography–mass spectrometry with matrix matching calibration strategy. Environ. Monit. Assess. 2022, 194, 58. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, M.; Gracia-Lor, E.; Sancho, J.V.; Hernandez, F. Importance of MS selectivity and chromatographic separation in LC-MS/MS-based methods when investigating pharmaceutical metabolites in water. Dipyrone as a case of study. J. Mass Spectrom. 2012, 47, 1040–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathod, R.H.; Chaudhari, S.R.; Patil, A.S.; Shirkhedkar, A.A. Ultra-high performance liquid chromatography-MS/MS (UHPLC-MS/MS) in practice: Analysis of drugs and pharmaceutical formulations. Future J. Pharm. Sci. 2019, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, K.; Tomoda, M.; Ohno, Y.; Hayashi, H.; Fujita, Y.; Kawakami, K.; Kameyama, K.; Kato, T.; Sugiyama, T.; Itoh, Y.; et al. Effects of Cabazitaxel in Renal Cell Carcinoma Cell Lines. Anticancer Res. 2015, 35, 6671–6677. [Google Scholar] [PubMed]
- European Commission. Commission Decision 2002/657/EC of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Communities 2002, 50, 8–36. [Google Scholar]
- ISO/IEC 17025: 2017. General Requirements for the Competence of Testing and Calibration Laboratories; International Organization for Standardization: Geneva, Switzerland, 2017.
- Caruso, R.; Scordino, M.; Traulo, P.; Gagliano, G. Determination of Volatile Compounds in Wine by Gas Chromatography-Flame Ionization Detection: Comparison Between the U.S. Environmental Protection Agency 3σ Approach and Hubaux-Vos Calculation of Detection Limits Using Ordinary and Bivariate Least Squares. J. AOAC Int. 2012, 95, 459–471. [Google Scholar] [CrossRef]
- Fekete, S.; Ganzler, K.; Fekete, J. Efficiency of the new sub-2 μm core–shell (Kinetex™) column in practice, applied for small and large molecule separation. J. Pharm. Biomed. Anal. 2011, 54, 482–490. [Google Scholar]
- Preti, R. Core-Shell Columns in High-Performance Liquid Chromatography: Food Analysis Applications. Int. J. Anal. Chem. 2016, 2016, 3189724. [Google Scholar] [CrossRef] [Green Version]
- Čelić, M.; Insa, S.; Škrbić, B.; Petrović, M. Development of a sensitive and robust online dual column liquid chromatography-tandem mass spectrometry method for the analysis of natural and synthetic estrogens and their conjugates in river water and wastewater. Anal. Bioanal. Chem. 2017, 409, 5427–5440. [Google Scholar] [CrossRef]
- Oliferova, L.; Statkus, M.; Tsysin, G.; Shpigun, O.; Zolotov, Y. On-line solid-phase extraction and HPLC determination of polycyclic aromatic hydrocarbons in water using fluorocarbon polymer sorbents. Anal. Chim. Acta 2005, 538, 35–40. [Google Scholar] [CrossRef]
- Garcia-Ac, A.; Segura, P.A.; Viglino, L.; Fürtös, A.; Gagnon, C.; Prévost, M.; Sauvé, S. On-line solid-phase extraction of large-volume injections coupled to liquid chromatography-tandem mass spectrometry for the quantitation and confirmation of 14 selected trace organic contaminants in drinking and surface water. J. Chromatogr. A 2009, 1216, 8518–8527. [Google Scholar] [CrossRef] [PubMed]
- Castiglioni, S.; Bagnati, R.; Calamari, D.; Fanelli, R.; Zuccato, E. A multiresidue analytical method using solid-phase extraction and high-pressure liquid chromatography tandem mass spectrometry to measure pharmaceuticals of different therapeutic classes in urban wastewaters. J. Chromatogr. A 2005, 1092, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Ray, N.M.; Wan, J.; Khan, A.; Chakraborty, T.; Ray, M.B. Micropollutants in Wastewater: Fate and Removal Processes. In Physico-Chemical Wastewater Treatment and Resource Recovery; IntechOpen: London, UK, 2017; p. 75. [Google Scholar] [CrossRef] [Green Version]
- Loos, R.; Carvalho, R.; António, D.C.; Comero, S.; Locoro, G.; Tavazzi, S.; Paracchini, B.; Ghiani, M.; Lettieri, T.; Blaha, L.; et al. EU-wide monitoring survey on emerging polar organic contaminants in wastewater treatment plant effluents. Water Res. 2013, 47, 6475–6487. [Google Scholar] [CrossRef] [PubMed]
- Ort, C.; Lawrence, M.G.; Reungoat, J.; Eaglesham, G.; Carter, S.; Keller, J. Determining the fraction of pharmaceutical residues in wastewater originating from a hospital. Water Res. 2010, 44, 605–615. [Google Scholar] [CrossRef]
- Feldmann, D.F.; Zuehlke, S.; Heberer, T. Occurrence, fate and assessment of polar metamizole (dipyrone) residues in hospital and municipal wastewater. Chemosphere 2008, 71, 1754–1764. [Google Scholar] [CrossRef]
- Weissbrodt, D.G.; Kovalova, L.; Ort, C.; Pazhepurackel, V.; Moser, R.; Hollender, J.; Siegrist, H.; McArdell, C.S. Mass Flows of X-ray Contrast Media and Cytostatics in Hospital Wastewater. Environ. Sci. Technol. 2009, 43, 4810–4817. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | Loading Pump (SPE) | Valve Position | Analytical Pump (HPLC) | |||
|---|---|---|---|---|---|---|
| A (%) | B (%) | Flow Rate (mL min−1) | C (%) | D (%) | ||
| 0.00 | 95.0 | 5.00 | 1.0 | Loading | 95.0 | 5.00 |
| 1.10 | 95.0 | 5.00 | 1.0 | Loading | 95.0 | 5.00 |
| 1.15 | 95.0 | 5.00 | 1.0 | Injection | 95.0 | 5.00 |
| 5.00 | 0.00 | 100 | 0.1 | Injection | 0.00 | 100 |
| 7.00 | 0.00 | 100 | 0.1 | Injection | 0.00 | 100 |
| 8.00 | 0.00 | 100 | 0.1 | Injection | 0.00 | 100 |
| 10.0 | 95.0 | 5.00 | 1.0 | Loading | 95.0 | 5.00 |
| 15.0 | 95.0 | 5.00 | 1.0 | Loading | 95.0 | 5.00 |
| Compound (Abbreviation) | Formula | RT (min) | Precursor Ion, [M + H]+ (m/z) | Product Ions (m/z) | Frag (V) | CE (V) |
|---|---|---|---|---|---|---|
| Aliskiren (ALK) | C30H53N3O6 | 5.0 | 552.4 | 346.3; 534.5 | 135 | 27; 40 |
| Cabazitaxel (CTX) | C45H57NO14 | 7.5 | 836.3 | 555.3; 433.1 | 135 | 20; 20 |
| Docetaxel (DTX) | C43H53NO14 | 6.5 | 808.3 | 527.1; 509.0 | 135 | 20; 15 |
| Doxorubicin (DOX) | C27H29NO11 | 6.1 | 544.0 | 361.2; 397.2 | 135 | 10; 10 |
| Etoposide (ETP) | C29H32O13 | 4.3 | 589.2 | 229.1; 185.2 | 135 | 15; 15 |
| Irinotecan (IRI) | C33H38N4O6 | 1.6 | 587.3 | 124.1; 167.1 | 120 | 45; 21 |
| Maprotiline (MAP) | C20H23N | 5.3 | 278.1 | 250.0; 191.1 | 135 | 15; 15 |
| Methotrexate (MTX) | C20H22N8O5 | 1.4 | 455.2 | 308.1; 175.1 | 120 | 10; 25 |
| Paclitaxel (PTX) | C47H51NO14 | 7.1 | 854.0 | 105.1; 286.0 | 100 | 19; 10 |
| Topotecan (TOP) | C23H23N3O5 | 3.8 | 422.2 | 377.1; 320.0 | 120 | 10; 21 |
| Atrazine-d5 (ATZ) | C8H5H9ClN5 | 5.6 | 221.1 | 179.2; 101.2 | 135 | 20; 20 |
| Compound | Linearity (R2) | LOD (ng L−1) | LOQ (ng L−1) | Spiked QC | Precision (RSD %) | Recovery (%) | |
|---|---|---|---|---|---|---|---|
| Intraday | Interday | ||||||
| ALK | 0.9978 | 10.7 | 35.5 | QCL | 5.6 | 13 | 101.6 (5.4) |
| QCM | 3.3 | 9.2 | 95.7 (10) | ||||
| QCH | 3.7 | 7.9 | 94.6 (2.0) | ||||
| CTX | 0.9937 | 7.98 | 26.6 | QCL | 5.5 | 7.0 | 101.3 (2.2) |
| QCM | 6.0 | 4.3 | 94.7 (7.5) | ||||
| QCH | 2.9 | 3.5 | 96.3 (7.1) | ||||
| DTX | 0.9987 | 2.67 | 8.89 | QCL | 7.8 | 11 | 84.0 (11) |
| QCM | 6.7 | 7.9 | 95.3 (8.9) | ||||
| QCH | 8.4 | 6.4 | 96.9 (7.8) | ||||
| DOX | 0.9978 | 2.27 | 7.57 | QCL | 6.1 | 7.5 | 80 (13) |
| QCM | 4.1 | 9.1 | 85.8 (2.9) | ||||
| QCH | 5.5 | 13 | 87.6 (9.1) | ||||
| ETP | 0.9947 | 3.25 | 10.9 | QCL | 3.7 | 9.6 | 104.5 (6.9) |
| QCM | 3.2 | 6.8 | 96.9 (3.8) | ||||
| QCH | 6.2 | 12 | 93.3 (6.8) | ||||
| IRI | 0.9975 | 7.96 | 26.5 | QCL | 4.8 | 9.3 | 78.4 (7.6) |
| QCM | 5.1 | 8.0 | 89.2 (5.4) | ||||
| QCH | 3.9 | 11 | 86.5 (4.4) | ||||
| MAP | 0.9997 | 1.30 | 4.34 | QCL | 2.6 | 8.5 | 103 (14) |
| QCM | 3.3 | 5.4 | 103.2 (6.4) | ||||
| QCH | 2.1 | 5.5 | 98.6 (8.2) | ||||
| MTX | 0.9991 | 4.43 | 14.8 | QCL | 2.1 | 10 | 85.4 (9.0) |
| QCM | 4.4 | 7.7 | 94.4 (5.6) | ||||
| QCH | 3.0 | 5.2 | 111.0 (6.4) | ||||
| PTX | 0.9969 | 6.99 | 23.3 | QCL | 6.6 | 7.8 | 96 (12) |
| QCM | 5.1 | 5.2 | 88.8 (9.2) | ||||
| QCH | 6.7 | 3.3 | 94.6 (7.4) | ||||
| TOP | 0.9982 | 4.22 | 14.1 | QCL | 7.5 | 12 | 92 (11) |
| QCM | 7.4 | 9.4 | 93.7 (9.1) | ||||
| QCH | 6.1 | 9.8 | 96.6 (7.4) | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belay, M.H.; Precht, U.; Mortensen, P.; Marengo, E.; Robotti, E. A Fully Automated Online SPE-LC-MS/MS Method for the Determination of 10 Pharmaceuticals in Wastewater Samples. Toxics 2022, 10, 103. https://doi.org/10.3390/toxics10030103
Belay MH, Precht U, Mortensen P, Marengo E, Robotti E. A Fully Automated Online SPE-LC-MS/MS Method for the Determination of 10 Pharmaceuticals in Wastewater Samples. Toxics. 2022; 10(3):103. https://doi.org/10.3390/toxics10030103
Chicago/Turabian StyleBelay, Masho Hilawie, Ulrich Precht, Peter Mortensen, Emilio Marengo, and Elisa Robotti. 2022. "A Fully Automated Online SPE-LC-MS/MS Method for the Determination of 10 Pharmaceuticals in Wastewater Samples" Toxics 10, no. 3: 103. https://doi.org/10.3390/toxics10030103