
L-Ascorbic Acid 2-Phosphate Attenuates Methylmercury-Induced Apoptosis by Inhibiting Reactive Oxygen Species Accumulation and DNA Damage in Human SH-SY5Y Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. SH-SY5Y Cell Culture
2.2. Treatment of SH-SY5Y Cells with MeHg and AA2P
2.3. Reactive Oxygen Species Assay
2.4. Apoptosis Assays
2.5. Alkaline Comet Assay
2.6. Western Blot Assay
2.7. Statistical Analysis
3. Results
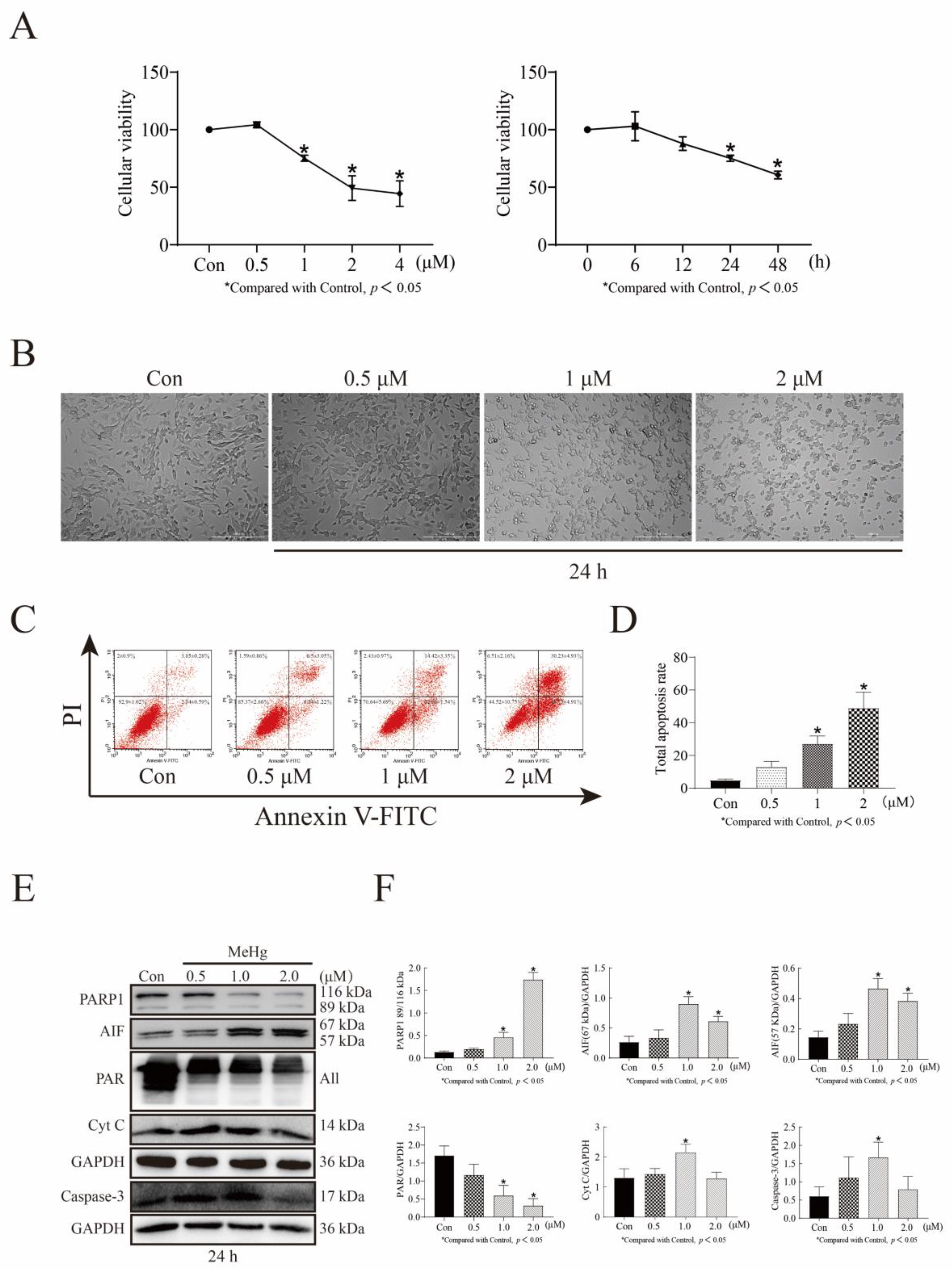
3.1. MeHg Decreases SH-SY5Y Cell Viability in a Time- and Dose-Dependent Manner and Increases Apoptosis in a Dose-Dependent Manner
3.2. MeHg Increases ROS Accumulation in SH-SY5Y Cells in a Time-Dependent Manner and Is Accompanied by an Increase in Apoptosis
3.3. MeHg Induces DNA-Damage-Mediated Repair and Caspase-Dependent and -Independent Apoptosis in SH-SY5Y Cells
3.4. AA2P Decreases MeHg-Induced ROS Accumulation in SH-5YSY Cells in a Dose-Dependent Manner
3.5. AA2P Decreases MeHg-Induced Apoptosis in SH-SY5Y Cells
3.6. AA2P Reduces MeHg-Induced DNA Damage and Repair in SH-SY5Y Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takahashi, T.; Shimohata, T. Vascular Dysfunction Induced by Mercury Exposure. Int. J. Mol. Sci. 2019, 20, 2435. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, M.; Itai, T.; Murata, K. Effects of Prenatal Methylmercury Exposure: From Minamata Disease to Environmental Health Studies. Nihon Eiseigaku Zasshi 2017, 72, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Santana, L.N.D.S.; Bittencourt, L.O.; Nascimento, P.C.; Fernandes, R.M.; Teixeira, F.B.; Fernandes, L.M.P.; Silva, M.C.F.; Nogueira, L.S.; Amado, L.L.; Crespo-Lopez, M.E.; et al. Low doses of methylmercury exposure during adulthood in rats display oxidative stress, neurodegeneration in the motor cortex and lead to impairment of motor skills. J. Trace Elem. Med. Biol. 2019, 51, 19–27. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, S.; Wang, Y.; Lu, A.; Hu, C.; Yan, C. DHA ameliorates MeHg-induced PC12 cell apoptosis by inhibiting the ROS/JNK signaling pathway. Mol. Med. Rep. 2021, 24, 558. [Google Scholar] [CrossRef] [PubMed]
- Bittencourt, L.O.; Dionizio, A.; Nascimento, P.C.; Puty, B.; Leão, L.K.R.; Luz, D.A.; Silva, M.C.F.; Amado, L.L.; Leite, A.; Buzalaf, M.R.; et al. Proteomic approach underlying the hippocampal neurodegeneration caused by low doses of methylmercury after long-term exposure in adult rats. Metallomics 2019, 11, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Abu Zeid, E.H.; Khalifa, B.A.; Said, E.N.; Arisha, A.H.; Reda, R.M. Neurobehavioral and immune-toxic impairments induced by organic methyl mercury dietary exposure in Nile tilapia Oreochromis niloticus. Aquat. Toxicol. 2021, 230, 105702. [Google Scholar] [CrossRef]
- Stringari, J.; Nunes, A.K.; Franco, J.L.; Bohrer, D.; Garcia, S.C.; Dafre, A.L.; Milatovic, D.; Souza, D.O.; Rocha, J.B.; Aschner, M.; et al. Prenatal methylmercury exposure hampers glutathione antioxidant system ontogenesis and causes long-lasting oxidative stress in the mouse brain. Toxicol. Appl. Pharmacol. 2008, 227, 147–154. [Google Scholar] [CrossRef]
- Franco, J.L.; Posser, T.; Dunkley, P.R.; Dickson, P.W.; Mattos, J.J.; Martins, R.; Bainy, A.C.; Marques, M.R.; Dafre, A.L.; Farina, M. Methylmercury neurotoxicity is associated with inhibition of the antioxidant enzyme glutathione peroxidase. Free. Radic. Biol. Med. 2009, 47, 449–457. [Google Scholar] [CrossRef]
- Chandran, A.M.K.; Christina, H.; DAS, S.; Mumbrekar, K.D.; Rao, B.S.S. Neuroprotective role of naringenin against methylmercury induced cognitive impairment and mitochondrial damage in a mouse model. Environ. Toxicol. Pharmacol. 2019, 71, 103224. [Google Scholar] [CrossRef]
- Bi, Y.; Lu, Z.; Wu, J.; Cheng, G.; Tian, J.; Lu, Z. Methylmercury chloride damage to the adult rat hippocampus cannot be detected by proton magnetic resonance spectroscopy. Neural Regen. Res. 2014, 9, 1616–1620. [Google Scholar] [CrossRef]
- do Nascimento, J.L.M.; Oliveira, K.R.M.; Crespo-Lopez, M.E.; Macchi, B.M.; Maués, L.A.; Pinheiro, M.D.C.N.; Silveira, L.C.L.; Herculano, A.M. Methylmercury neurotoxicity & antioxidant defenses. Indian J. Med. Res. 2008, 128, 373–382. [Google Scholar] [PubMed]
- Patel, E.; Reynolds, M. Methylmercury impairs motor function in early development and induces oxidative stress in cerebellar granule cells. Toxicol. Lett. 2013, 222, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Caballero, B.; Olguin, N.; Campos, F.; Farina, M.; Ballester, F.; Lopez-Espinosa, M.-J.; Llop, S.; Rodríguez-Farré, E.; Suñol, C. Methylmercury-induced developmental toxicity is associated with oxidative stress and cofilin phosphorylation. Cellular and human studies. Neurotoxicology 2017, 59, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Xu, Z.; Liu, W.; Xu, B.; Deng, Y. Oxidative stress accelerates synaptic glutamate dyshomeostasis and NMDARs disorder during methylmercury-induced neuronal apoptosis in rat cerebral cortex. Environ. Toxicol. 2020, 35, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Yin, C.; Zhou, Y.; Wang, Q.; Jiang, Y.; Bai, Y.; Qian, H.; Xing, G.; Wang, S.; Li, F.; et al. Curcumin protects against methylmercury-induced cytotoxicity in primary rat astrocytes by activating the Nrf2/ARE pathway independently of PKCδ. Toxicology 2019, 425, 152248. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Choi, S.-H.; Lee, N.-E.; Cho, H.-J.; Rhim, H.; Kim, H.-C.; Hwang, S.-H.; Nah, S.-Y. Effects of Gintonin-Enriched Fraction on Methylmercury-Induced Neurotoxicity and Organ Methylmercury Elimination. Int. J. Environ. Res. Public Health 2020, 17, 838. [Google Scholar] [CrossRef]
- Joshi, D.; Kumar, M.D.; Kumar, S.A.; Sangeeta, S. Reversal of Methylmercury-Induced Oxidative Stress, Lipid Peroxidation, and DNA Damage by the Treatment of N-Acetyl Cysteine: A Protective Approach. J. Environ. Pathol. Toxicol. Oncol. 2014, 33, 167–182. [Google Scholar] [CrossRef]
- Yin, Z.; Lee, E.; Ni, M.; Jiang, H.; Milatovic, D.; Rongzhu, L.; Farina, M.; da Rocha, J.B.T.; Aschner, M. Methylmercury-induced alterations in astrocyte functions are attenuated by ebselen. Neurotoxicology 2011, 32, 291–299. [Google Scholar] [CrossRef]
- Feng, S.; Xu, Z.; Wang, F.; Yang, T.; Liu, W.; Deng, Y.; Xu, B. Sulforaphane Prevents Methylmercury-Induced Oxidative Damage and Excitotoxicity Through Activation of the Nrf2-ARE Pathway. Mol. Neurobiol. 2017, 54, 375–391. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Yumvihoze, E.; Chan, H.M. Superoxide anion radical (O2−) degrades methylmercury to inorganic mercury in human astrocytoma cell line (CCF-STTG1). Chem. Biol. Interact. 2015, 239, 46–55. [Google Scholar] [CrossRef]
- Das, S.; Paul, A.; Mumbrekar, K.D.; Rao, S.B.S. Harmonization of Mangiferin on methylmercury engendered mitochondrial dysfunction. Environ. Toxicol. 2017, 32, 630–644. [Google Scholar] [CrossRef]
- Ishihara, Y.; Tsuji, M.; Kawamoto, T.; Yamazaki, T. Involvement of reactive oxygen species derived from mitochondria in neuronal injury elicited by methylmercury. J. Clin. Biochem. Nutr. 2016, 59, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.F.; Ammar, D.; Bourckhardt, G.F.; Kobus-Bianchini, K.; Müller, Y.M.R.; Nazari, E.M. MeHg Developing Exposure Causes DNA Double-Strand Breaks and Elicits Cell Cycle Arrest in Spinal Cord Cells. J. Toxicol. 2015, 2015, 532691. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, Y.; Yan, X.-P. Probing Mercury Species−DNA Interactions by Capillary Electrophoresis with On-Line Electrothermal Atomic Absorption Spectrometric Detection. Anal. Chem. 2006, 78, 6115–6120. [Google Scholar] [CrossRef] [PubMed]
- Belletti, S.; Orlandini, G.; Vettori, M.V.; Mutti, A.; Uggeri, J.; Scandroglio, R.; Alinovi, R.; Gatti, R. Time course assessment of methylmercury effects on C6 glioma cells: Submicromolar concentrations induce oxidative DNA damage and apoptosis. J. Neurosci. Res. 2002, 70, 703–711. [Google Scholar] [CrossRef]
- Liu, W.; Yang, T.; Xu, Z.; Xu, B.; Deng, Y. Methyl-mercury induces apoptosis through ROS-mediated endoplasmic reticulum stress and mitochondrial apoptosis pathways activation in rat cortical neurons. Free. Radic. Res. 2018, 53, 26–44. [Google Scholar] [CrossRef] [PubMed]
- Qu, M.; Nan, X.; Gao, Z.; Guo, B.; Liu, B.; Chen, Z. Protective effects of lycopene against methylmercury-induced neurotoxicity in cultured rat cerebellar granule neurons. Brain Res. 2013, 1540, 92–102. [Google Scholar] [CrossRef]
- Roos, D.; Seeger, R.; Puntel, R.; Barbosa, N.V. Role of Calcium and Mitochondria in MeHg-Mediated Cytotoxicity. J. Biomed. Biotechnol. 2012, 2012, 248764. [Google Scholar] [CrossRef]
- Tamm, C.; Duckworth, J.; Hermanson, O.; Ceccatelli, S. High susceptibility of neural stem cells to methylmercury toxicity: Effects on cell survival and neuronal differentiation. J. Neurochem. 2006, 97, 69–78. [Google Scholar] [CrossRef]
- dos Santos, A.A.; López-Granero, C.; Farina, M.; Rocha, J.B.; Bowman, A.B.; Aschner, M. Oxidative stress, caspase-3 activation and cleavage of ROCK-1 play an essential role in MeHg-induced cell death in primary astroglial cells. Food Chem. Toxicol. 2018, 113, 328–336. [Google Scholar] [CrossRef]
- Bratton, S.B.; MacFarlane, M.; Cain, K.; Cohen, G.M. Protein Complexes Activate Distinct Caspase Cascades in Death Receptor and Stress-Induced Apoptosis. Exp. Cell Res. 2000, 256, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Carocci, A.; Rovito, N.; Sinicropi, M.S.; Genchi, G. Mercury Toxicity and Neurodegenerative Effects. Rev. Environ. Contam. Toxicol. 2014, 229, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Fonfría, E.; Daré, E.; Benelli, M.; Suñol, C.; Ceccatelli, S. Translocation of apoptosis-inducing factor in cerebellar granule cells exposed to neurotoxic agents inducing oxidative stress. Eur. J. Neurosci. 2002, 16, 2013–2016. [Google Scholar] [CrossRef] [PubMed]
- Slagsvold, H.H.; Rosseland, C.M.; Jacobs, C.; Khuong, E.; Kristoffersen, N.; Gaarder, M.; Fallgren, A.B.; Huitfeldt, H.S.; Paulsen, R.E. High molecular weight DNA fragments are processed by caspase sensitive or caspase independent pathways in cultures of cerebellar granule neurons. Brain Res. 2003, 984, 111–121. [Google Scholar] [CrossRef]
- Chung, Y.-P.; Yen, C.-C.; Tang, F.-C.; Lee, K.-I.; Liu, S.-H.; Wu, C.-C.; Hsieh, S.-S.; Su, C.-C.; Kuo, C.-Y.; Chen, Y.-W. Methylmercury exposure induces ROS/Akt inactivation-triggered endoplasmic reticulum stress-regulated neuronal cell apoptosis. Toxicology 2019, 425, 152245. [Google Scholar] [CrossRef]
- Hou, S.; Zhang, X.; Ning, X.; Wu, H.; Li, X.; Ma, K.; Hao, H.; Lv, C.; Li, C.; Du, Z.; et al. Methylmercury induced apoptosis of human neuroblastoma cells through the reactive oxygen species mediated caspase and poly ADP-ribose polymerase/ a poptosis-inducing factor dependent pathways. Environ. Toxicol. 2022, 37, 1891–1901. [Google Scholar] [CrossRef]
- Kocot, J.; Luchowska-Kocot, D.; Kiełczykowska, M.; Musik, I.; Kurzepa, J. Does Vitamin C Influence Neurodegenerative Diseases and Psychiatric Disorders? Nutrients 2017, 9, 659. [Google Scholar] [CrossRef]
- Špiclin, P.; Homar, M.; Zupančič-Valant, A.; Gašperlin, M. Sodium ascorbyl phosphate in topical microemulsions. Int. J. Pharm. 2003, 256, 65–73. [Google Scholar] [CrossRef]
- Monacelli, F.; Acquarone, F.M.E.; Giannotti, C.; Borghi, R.; Nencioni, A. Vitamin C, Aging and Alzheimer’s Disease. Nutrients 2017, 9, 670. [Google Scholar] [CrossRef]
- Carr, A.; Frei, B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J. 1999, 13, 1007–1024. [Google Scholar] [CrossRef] [Green Version]
- Kiyatkin, E.A.; Rebec, G.V. Ascorbate modulates glutamate-induced excitations of striatal neurons. Brain Res. 1998, 812, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Méndez, R.; Rivas-Arancibia, S. Vitamin C in Health and Disease: Its Role in the Metabolism of Cells and Redox State in the Brain. Front. Physiol. 2015, 6, 397. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.-H.; Chen, S.; Corpe, C.; Dutta, A.; Dutta, S.K.; et al. Vitamin C as an Antioxidant: Evaluation of Its Role in Disease Prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Chang, M.-Y.; Park, C.-H.; Kim, H.-Y.; Kim, J.-H.; Son, H.; Lee, Y.-S.; Lee, S.-H. Ascorbate-induced differentiation of embryonic cortical precursors into neurons and astrocytes. J. Neurosci. Res. 2003, 73, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Parle, M.; Dhingra, D. Ascorbic Acid: A Promising Memory-Enhancer in Mice. J. Pharmacol. Sci. 2003, 93, 129–135. [Google Scholar] [CrossRef]
- Puty, B.; Maximino, C.; Brasil, A.; da Silva, W.L.L.; Gouveia, A., Jr.; Oliveira, K.R.M.; Batista, E.D.J.O.; Crespo-Lopez, M.E.; Rocha, F.A.F.; Herculano, A.M. Ascorbic Acid Protects against Anxiogenic-Like Effect Induced by Methylmercury in Zebrafish: Action on the Serotonergic System. Zebrafish 2014, 11, 365–370. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Lee, S.; Park, Y.; Min, T.; Bai, S.C. Evaluation of dietary selenium, vitamin C and E as the multi-antioxidants on the methylmercury intoxicated mice based on mercury bioaccumulation, antioxidant enzyme activity, lipid peroxidation and mitochondrial oxidative stress. Chemosphere 2021, 273, 129673. [Google Scholar] [CrossRef]
- Konopacka, M.; Palyvoda, O.; Rzeszowska-Wolny, J. Inhibitory effect of ascorbic acid post-treatment on radiation-induced chromosomal damage in human lymphocytes in vitro. Teratog. Carcinog. Mutagen. 2002, 22, 443–450. [Google Scholar] [CrossRef]
- Kawashima, S.; Funakoshi, T.; Sato, Y.; Saito, N.; Ohsawa, H.; Kurita, K.; Nagata, K.; Yoshida, M.; Ishigami, A. Protective effect of pre- and post-vitamin C treatments on UVB-irradiation-induced skin damage. Sci. Rep. 2018, 8, 16199. [Google Scholar] [CrossRef]
- Olguín, N.; Müller, M.-L.; Rodríguez-Farré, E.; Suñol, C. Neurotransmitter amines and antioxidant agents in neuronal protection against methylmercury-induced cytotoxicity in primary cultures of mice cortical neurons. Neurotoxicology 2018, 69, 278–287. [Google Scholar] [CrossRef]
- Farina, M.; Aschner, M. Methylmercury-Induced Neurotoxicity: Focus on Pro-oxidative Events and Related Consequences. Adv. Neurobiol. 2017, 18, 267–286. [Google Scholar] [CrossRef] [PubMed]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, T.; Zhang, H.; Gou, X.; Han, C.; Wang, J.; Chen, A.T.; Ma, J.; Liu, J.; Chen, Z.; et al. LRRC31 inhibits DNA repair and sensitizes breast cancer brain metastasis to radiation therapy. Nat. Cell Biol. 2020, 22, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Foo, T.K.; Xia, B. BRCA1-Dependent and Independent Recruitment of PALB2–BRCA2–RAD51 in the DNA Damage Response and Cancer. Cancer Res. 2022, 82, 3191–3197. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yu, X. The role of poly(ADP-ribosyl)ation in DNA damage response and cancer chemotherapy. Oncogene 2015, 34, 3349–3356. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.; Dominique, Y.; Massabuau, J.C.; Boudou, A.; Bourdineaud, J.P. Comparative Effects of Dietary Methylmercury on Gene Expression in Liver, Skeletal Muscle, and Brain of the Zebrafish (Danio rerio). Environ. Sci. Technol. 2005, 39, 3972–3980. [Google Scholar] [CrossRef]
- Cameron, A.M.; Castoldi, A.; Sanin, D.E.; Flachsmann, J.L.; Field, C.S.; Puleston, D.J.; Kyle, R.L.; Patterson, A.E.; Hässler, F.; Buescher, J.M.; et al. Inflammatory macrophage dependence on NAD+ salvage is a consequence of reactive oxygen species–mediated DNA damage. Nat. Immunol. 2019, 20, 420–432. [Google Scholar] [CrossRef]
- Caron, M.-C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.-P.; Langelier, M.-F.; et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 2019, 10, 2954. [Google Scholar] [CrossRef]
- Pan, Y.-R.; Song, J.-Y.; Fan, B.; Wang, Y.; Che, L.; Zhang, S.-M.; Chang, Y.-X.; He, C.; Li, G.-Y. mTOR may interact with PARP-1 to regulate visible light-induced parthanatos in photoreceptors. Cell Commun. Signal. 2020, 18, 27. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. Nitric Oxide Signaling in Neurodegeneration and Cell Death. Adv. Pharmacol. 2018, 82, 57–83. [Google Scholar]
- Ishii, T.; Igawa, T.; Hayakawa, H.; Fujita, T.; Sekiguchi, M.; Nakabeppu, Y. PCBP1 and PCBP2 both bind heavily oxidized RNA but cause opposing outcomes, suppressing or increasing apoptosis under oxidative conditions. J. Biol. Chem. 2020, 295, 12247–12261. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, M.; Onishi, M.; Uno, A.; Tanimichi, A.; Nobeyama, A.; Mori, M.; Yamada, S.; Negi, S.; Bu, X.; Kato, J.; et al. The 89-kDa PARP1 cleavage fragment serves as a cytoplasmic PAR carrier to induce AIF-mediated apoptosis. J. Biol. Chem. 2021, 296, 100046. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.L.; Lu, J.; Zhang, X.; Arnér, E.S.J.; Holmgren, A. Effects of selenite and chelating agents on mammalian thioredoxin reductase inhibited by mercury: Implications for treatment of mercury poisoning. FASEB J. 2011, 25, 370–381. [Google Scholar] [CrossRef]
- Mir, H.A.; Ali, R.; Wani, Z.A.; Khanday, F.A. Pro-oxidant vitamin C mechanistically exploits p66Shc/Rac1 GTPase pathway in inducing cytotoxicity. Int. J. Biol. Macromol. 2022, 205, 154–168. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuo, K.; Xu, Q.; Wang, Y.; Sui, Y.; Niu, Y.; Liu, Z.; Liu, M.; Liu, X.; Liu, D.; Sun, W.; et al. L-Ascorbic Acid 2-Phosphate Attenuates Methylmercury-Induced Apoptosis by Inhibiting Reactive Oxygen Species Accumulation and DNA Damage in Human SH-SY5Y Cells. Toxics 2023, 11, 144. https://doi.org/10.3390/toxics11020144
Zuo K, Xu Q, Wang Y, Sui Y, Niu Y, Liu Z, Liu M, Liu X, Liu D, Sun W, et al. L-Ascorbic Acid 2-Phosphate Attenuates Methylmercury-Induced Apoptosis by Inhibiting Reactive Oxygen Species Accumulation and DNA Damage in Human SH-SY5Y Cells. Toxics. 2023; 11(2):144. https://doi.org/10.3390/toxics11020144
Chicago/Turabian StyleZuo, Kuiyang, Qi Xu, Yujie Wang, Yutong Sui, Ye Niu, Zinan Liu, Mingsheng Liu, Xinpeng Liu, Dan Liu, Wei Sun, and et al. 2023. "L-Ascorbic Acid 2-Phosphate Attenuates Methylmercury-Induced Apoptosis by Inhibiting Reactive Oxygen Species Accumulation and DNA Damage in Human SH-SY5Y Cells" Toxics 11, no. 2: 144. https://doi.org/10.3390/toxics11020144