
Mitochondrial Toxic Effects of Antiepileptic Drug Valproic Acid on Mouse Kidney Stem Cells




Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Measurement of Cell Viability
2.3. Measurement of mtROS
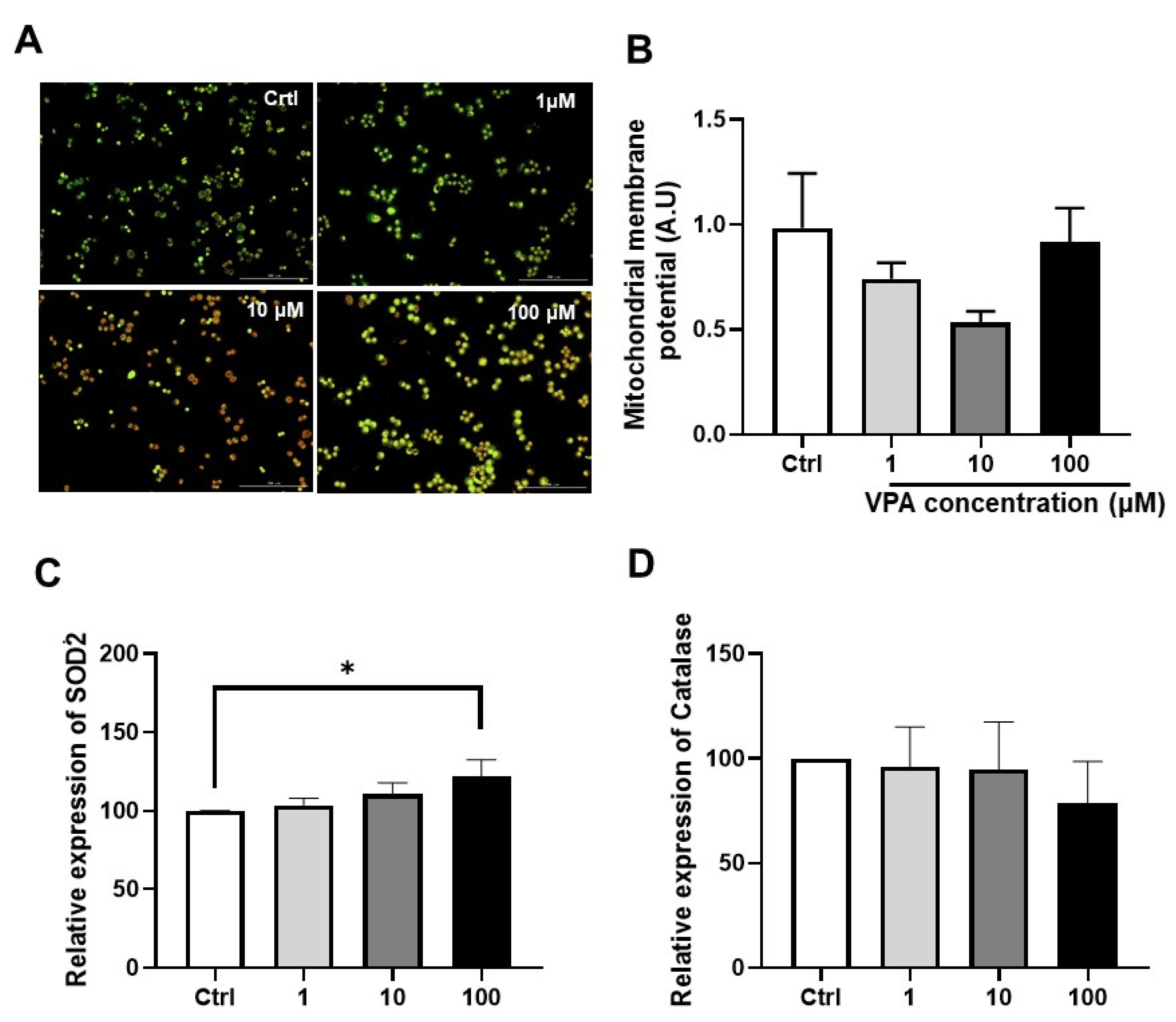
2.4. Measurement of Mitochondrial Membrane Potential
2.5. RNA Purification, cDNA Synthesis, and RT-PCR
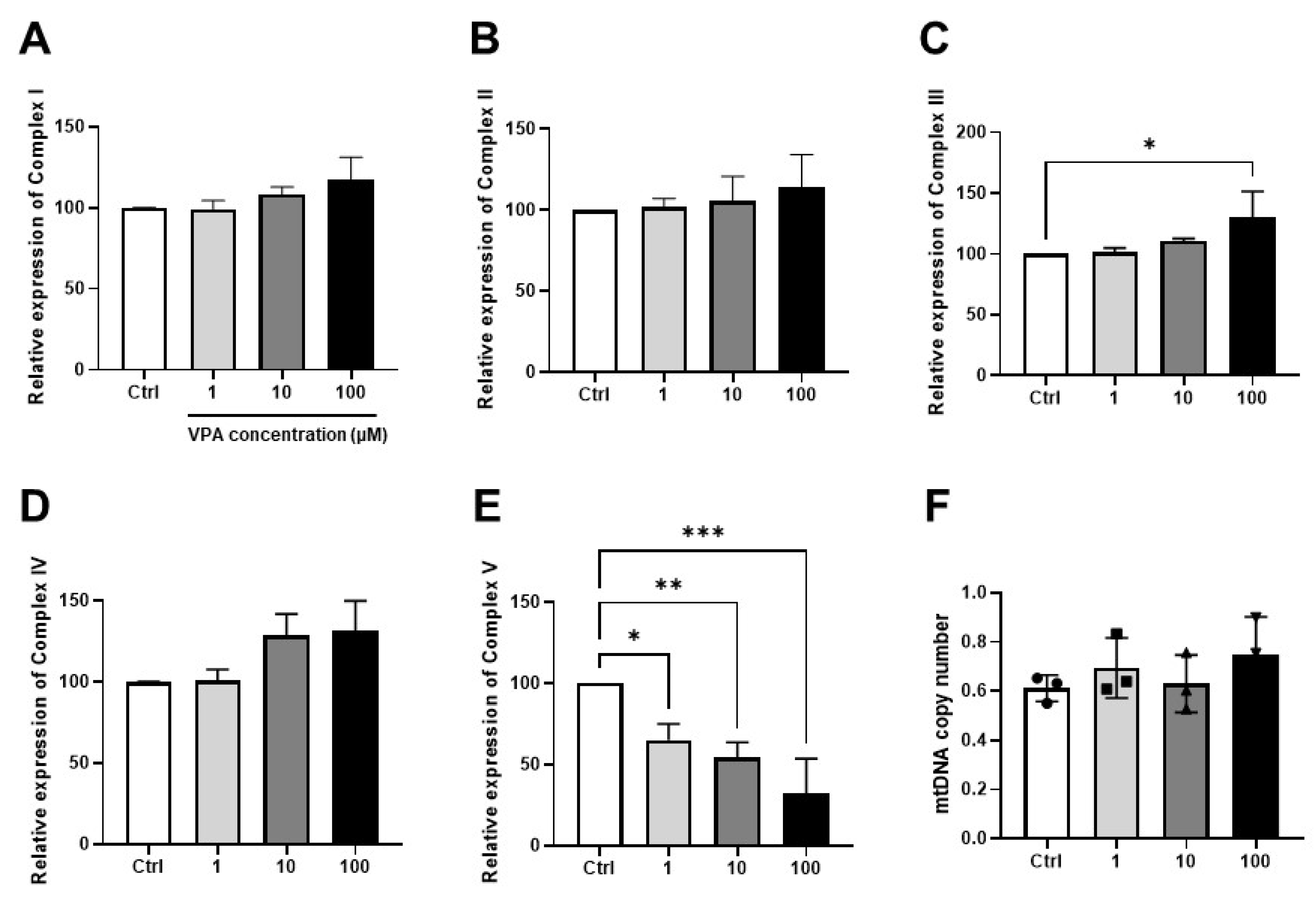
2.6. Analysis of Mitochondrial Gene Expression and mtDNA Copy Number
2.7. Statistical Analysis
3. Results
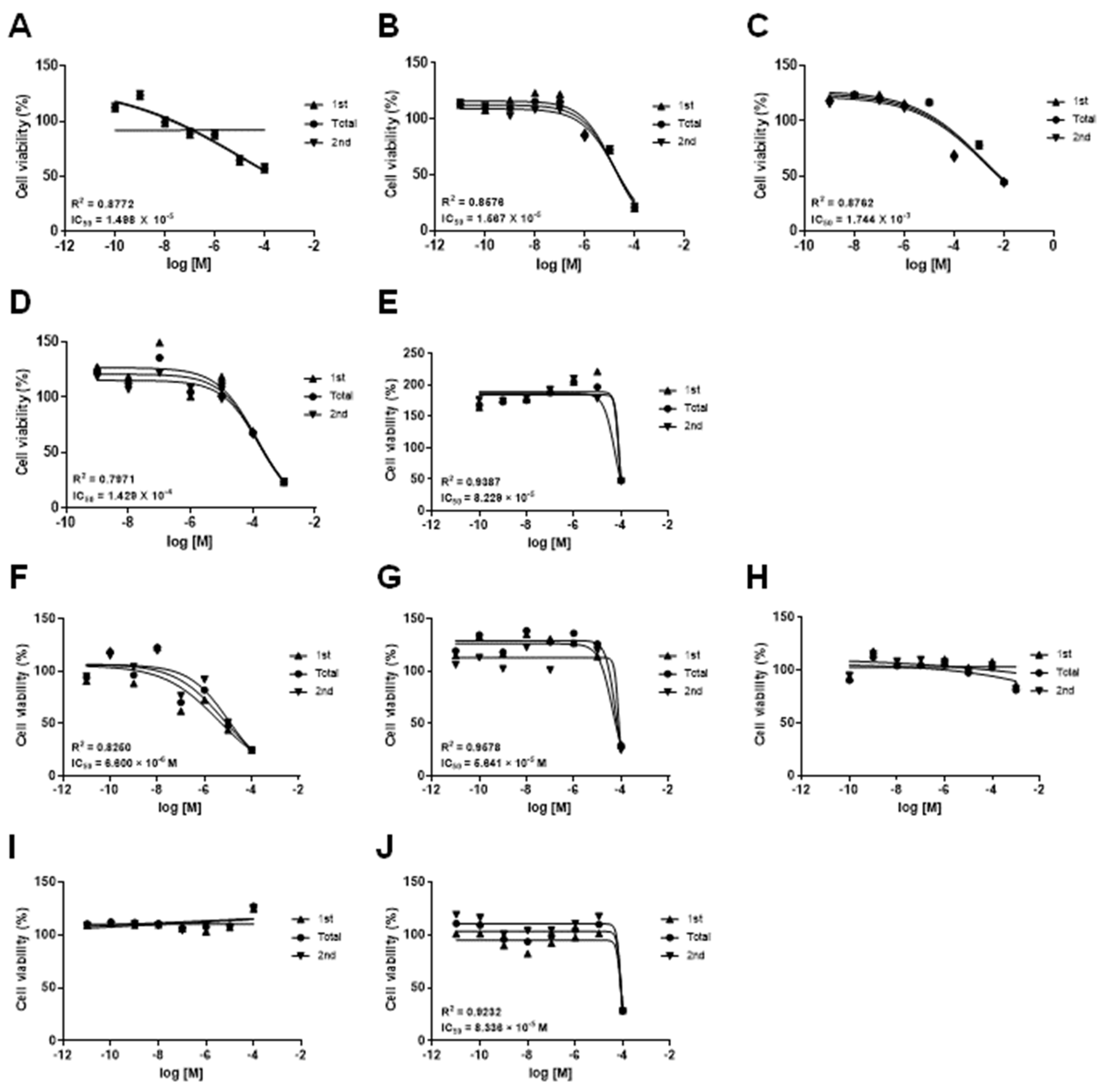
3.1. Effects of Drugs on the mKSCs and HEK293F Cell Viability
3.2. Effect of Drug on Mitochondrial Function in mKSCs and HEK293F
3.3. Effect of VPA on Mitochondrial Contents in mKSCs
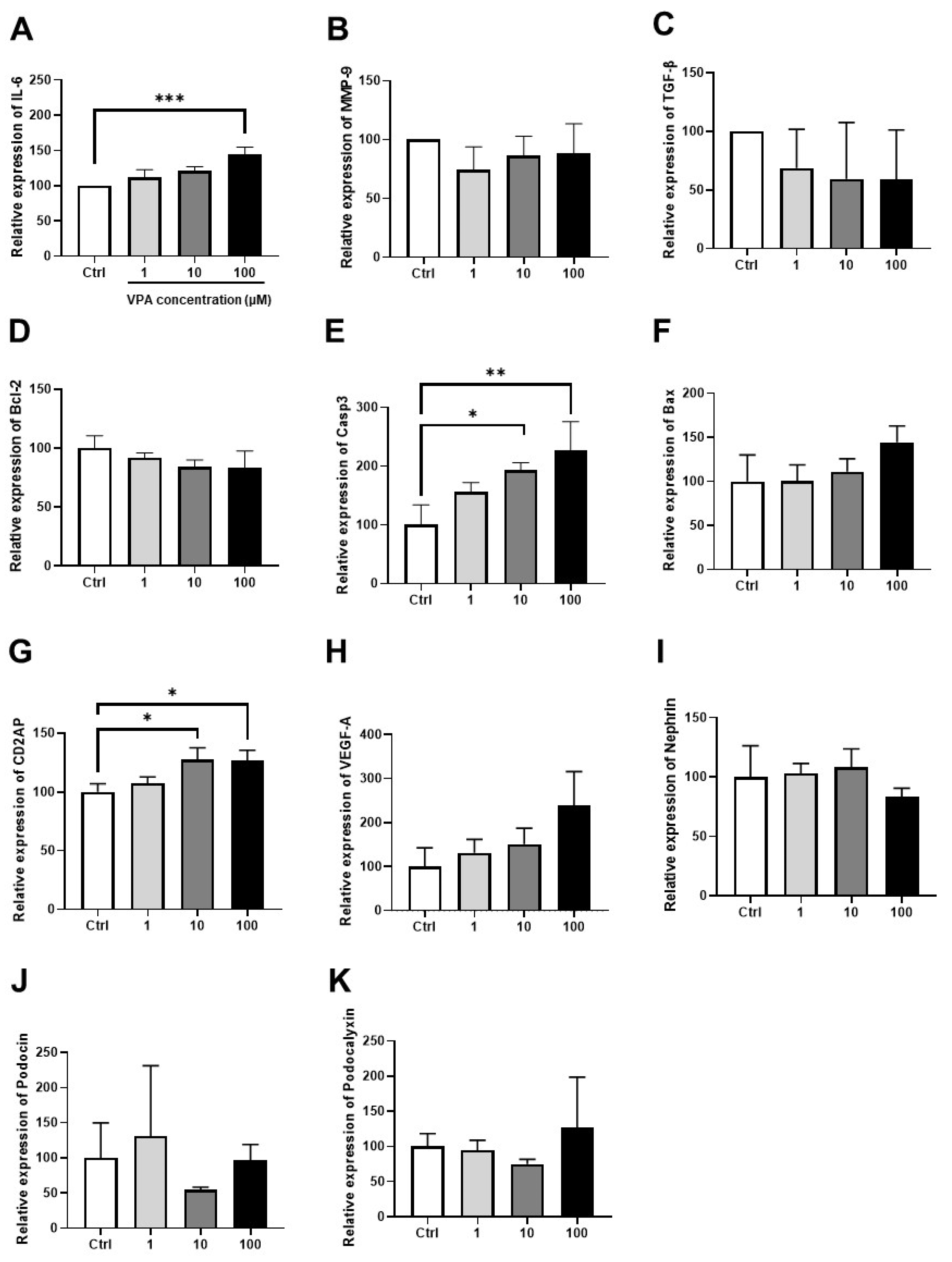
3.4. Effect of VPA on mKSCs Inflammation, Apoptosis, and Podocyte Injury Marker
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gravemann, U.; Volland, J.; Nau, H. Hydroxamic acid and fluorinated derivatives of valproic acid: Anticonvulsant activity, neurotoxicity and teratogenicity. Neurotoxicol. Teratol. 2008, 30, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Ghodke-Puranik, Y.; Thorn, C.F.; Lamba, J.K.; Leeder, J.S.; Song, W.; Birnbaum, A.K.; Altman, R.B.; Klein, T.E. Valproic acid pathway: Pharmacokinetics and pharmacodynamics. Pharm. Genom. 2013, 23, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Massart, J.; Robin, M.-A.; Borgne-Sanchez, A.; Fromenty, B. Drug-induced toxicity on mitochondria and lipid metabolism: Mechanistic diversity and deleterious consequences for the liver. J. Hepatol. 2011, 54, 773–794. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, A.; Rahman, J.; Banerjee, D.; Hakim Mohammed, A.R.; Malik, B.H. Parkinsonism: A Rare Adverse Effect of Valproic Acid. Cureus 2020, 12, e8782. [Google Scholar] [CrossRef]
- Abbate, M.; Zoja, C.; Remuzzi, G. How does proteinuria cause progressive renal damage? J. Am. Soc. Nephrol. 2006, 17, 2974–2984. [Google Scholar] [CrossRef]
- El-Shenawy, N.S.; Hamza, R.Z. Nephrotoxicity of sodium valproate and protective role of L-cysteine in rats at biochemical and histological levels. J. Basic Clin. Physiol. Pharmacol. 2016, 27, 497–504. [Google Scholar] [CrossRef]
- Costalonga, E.C.; Silva, F.M.; Noronha, I.L. Valproic Acid Prevents Renal Dysfunction and Inflammation in the Ischemia-Reperfusion Injury Model. BioMed Res. Int. 2016, 2016, 5985903. [Google Scholar] [CrossRef]
- Faria, J.; Ahmed, S.; Gerritsen, K.G.F.; Mihaila, S.M.; Masereeuw, R. Kidney-based in vitro models for drug-induced toxicity testing. Arch. Toxicol. 2019, 93, 3397–3418. [Google Scholar] [CrossRef]
- Chuah, J.K.C.; Zink, D. Stem cell-derived kidney cells and organoids: Recent breakthroughs and emerging applications. Biotechnol. Adv. 2017, 35, 150–167. [Google Scholar] [CrossRef]
- Fedotcheva, T.A.; Fedotcheva, N.I. Protectors of the Mitochondrial Permeability Transition Pore Activated by Iron and Doxorubicin. Curr. Cancer Drug Targets 2021, 21, 514–525. [Google Scholar] [CrossRef]
- Cardoso, C.M.; Almeida, L.M.; Custodio, J.B. 4-Hydroxytamoxifen is a potent inhibitor of the mitochondrial permeability transition. Mitochondrion 2002, 1, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Kon, K.; Kim, J.S.; Jaeschke, H.; Lemasters, J.J. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 2004, 40, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Guo, J.; Ying, Z.; Chen, S.; Yang, L.; Chen, K.; Long, Q.; Qin, D.; Pei, D.; Liu, X. Valproic acid-induced hepatotoxicity in Alpers syndrome is associated with mitochondrial permeability transition pore opening-dependent apoptotic sensitivity in an induced pluripotent stem cell model. Hepatology 2015, 61, 1730–1739. [Google Scholar] [CrossRef]
- Varbiro, G.; Toth, A.; Tapodi, A.; Bognar, Z.; Veres, B.; Sumegi, B.; Gallyas, F., Jr. Protective effect of amiodarone but not N-desethylamiodarone on postischemic hearts through the inhibition of mitochondrial permeability transition. J. Pharmacol. Exp. Ther. 2003, 307, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Daehn, I.; Casalena, G.; Zhang, T.; Shi, S.; Fenninger, F.; Barasch, N.; Yu, L.; D’Agati, V.; Schlondorff, D.; Kriz, W.; et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J. Clin. Investig. 2014, 124, 1608–1621. [Google Scholar] [CrossRef]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef]
- Markowitz, G.S.; Fine, P.L.; Stack, J.I.; Kunis, C.L.; Radhakrishnan, J.; Palecki, W.; Park, J.; Nasr, S.H.; Hoh, S.; Siegel, D.S.; et al. Toxic acute tubular necrosis following treatment with zoledronate (Zometa). Kidney Int. 2003, 64, 281–289. [Google Scholar] [CrossRef]
- Bae, E.; Lee, T.W.; Park, D.J. Drug-induced nephrotoxicity. JKMA 2020, 63, 30–35. [Google Scholar] [CrossRef]
- Perazella, M.A. Drug-induced nephropathy: An update. Expert Opin. Drug Saf. 2005, 4, 689–706. [Google Scholar] [CrossRef]
- Ramesh, G.; Ranganathan, P. Mouse models and methods for studying human disease, acute kidney injury (AKI). Methods Mol. Biol. 2014, 1194, 421–436. [Google Scholar] [CrossRef]
- Funk, J.A.; Schnellmann, R.G. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am. J. Physiol. Renal Physiol. 2012, 302, F853–F864. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Briones-Herrera, A.; Tapia, E.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective effects of N-acetyl-cysteine in mitochondria bioenergetics, oxidative stress, dynamics and S-glutathionylation alterations in acute kidney damage induced by folic acid. Free Radic. Biol. Med. 2019, 130, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Menzies, K.J.; Auwerx, J. The role of mitochondria in stem cell fate and aging. Development 2018, 145, dev143420. [Google Scholar] [CrossRef] [PubMed]
- Sitarz, K.S.; Elliott, H.R.; Karaman, B.S.; Relton, C.; Chinnery, P.F.; Horvath, R. Valproic acid triggers increased mitochondrial biogenesis in POLG-deficient fibroblasts. Mol. Genet. Metab. 2014, 112, 57–63. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144. [Google Scholar] [CrossRef]
- Chan, K.; Truong, D.; Shangari, N.; O’Brien, P.J. Drug-induced mitochondrial toxicity. Expert Opin. Drug Metab. Toxicol. 2005, 1, 655–669. [Google Scholar] [CrossRef]
- Silva, J.P.; Shabalina, I.G.; Dufour, E.; Petrovic, N.; Backlund, E.C.; Hultenby, K.; Wibom, R.; Nedergaard, J.; Cannon, B.; Larsson, N.G. SOD2 overexpression: Enhanced mitochondrial tolerance but absence of effect on UCP activity. EMBO J. 2005, 24, 4061–4070. [Google Scholar] [CrossRef] [PubMed]
- Salsaa, M.; Pereira, B.; Liu, J.; Yu, W.; Jadhav, S.; Hüttemann, M.; Greenberg, M.L. Valproate inhibits mitochondrial bioenergetics and increases glycolysis in Saccharomyces cerevisiae. Sci. Rep. 2020, 10, 11785. [Google Scholar] [CrossRef]
- Sousa, J.S.; D’Imprima, E.; Vonck, J. Mitochondrial Respiratory Chain Complexes. In Membrane Protein Complexes: Structure and Function; Harris, J.R., Boekema, E.J., Eds.; Springer: Singapore, 2018; pp. 167–227. [Google Scholar] [CrossRef]
- Jian, Z.; Guo, H.; Liu, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Oxidative stress, apoptosis and inflammatory responses involved in copper-induced pulmonary toxicity in mice. Aging 2020, 12, 16867–16886. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Lee, E.Y.; Lee, M.Y.; Hong, S.W.; Chung, C.H.; Hong, S.Y. Blockade of oxidative stress by vitamin C ameliorates albuminuria and renal sclerosis in experimental diabetic rats. Yonsei Med. J. 2007, 48, 847–855. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | CAS No. | Source | IC50 (M) | |
|---|---|---|---|---|
| ABC-SC0037 | HEK293F | |||
| Doxorubicin hydrochloride | 25316-40-9 | Sigma-Aldrich, MA, USA | 1.498 × 10−5 | 6.600 × 10−6 |
| (Z)-4-Hydroxytamoxifen | 68047-06-3 | Tocris, Bristol, UK | 1.567 × 10−5 | 5.641 × 10−5 |
| Amiodarone hydrochloride | 19774-82-4 | Sigma-Aldrich | 8.229 × 10−5 | 8.336 × 10−5 |
| Acetaminophen | 103-90-2 | Sigma-Aldrich | 1.744 × 10−3 | - |
| Valproic acid sodium salt | 1069-66-5 | Sigma-Aldrich | 1.429 × 10−4 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.; Ahn, C.; Kim, K.; Jeung, E.-B. Mitochondrial Toxic Effects of Antiepileptic Drug Valproic Acid on Mouse Kidney Stem Cells. Toxics 2023, 11, 471. https://doi.org/10.3390/toxics11050471
Lee M, Ahn C, Kim K, Jeung E-B. Mitochondrial Toxic Effects of Antiepileptic Drug Valproic Acid on Mouse Kidney Stem Cells. Toxics. 2023; 11(5):471. https://doi.org/10.3390/toxics11050471
Chicago/Turabian StyleLee, Minsu, Changhwan Ahn, KangMin Kim, and Eui-Bae Jeung. 2023. "Mitochondrial Toxic Effects of Antiepileptic Drug Valproic Acid on Mouse Kidney Stem Cells" Toxics 11, no. 5: 471. https://doi.org/10.3390/toxics11050471
APA StyleLee, M., Ahn, C., Kim, K., & Jeung, E.-B. (2023). Mitochondrial Toxic Effects of Antiepileptic Drug Valproic Acid on Mouse Kidney Stem Cells. Toxics, 11(5), 471. https://doi.org/10.3390/toxics11050471