
A Multi-Omics Study of Neurodamage Induced by Growth-Stage Real-Time Air Pollution Exposure in Mice via the Microbiome–Gut–Brain Axis


 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
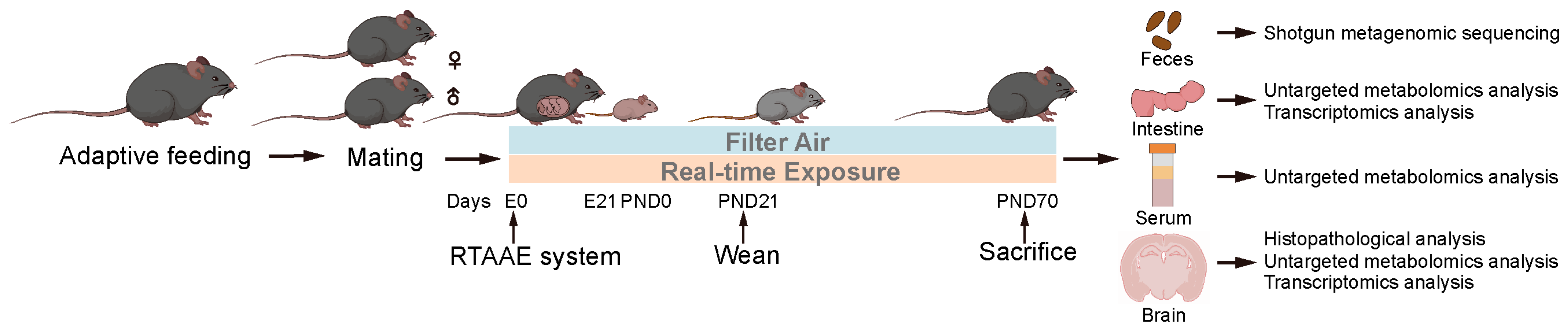
2.1. Animals and Study Design
2.2. Real-Time Ambient Air Pollution Exposure (RTAAE) System
2.3. Histopathological Analysis
2.4. Gut Microbiome Assessment by Shotgun Metagenomic Sequencing
2.5. Untargeted Metabolomics Analysis
2.6. Transcriptomics Analysis
2.7. Statistical Analysis
3. Results
3.1. Level of Air Pollutants in the Exposure System
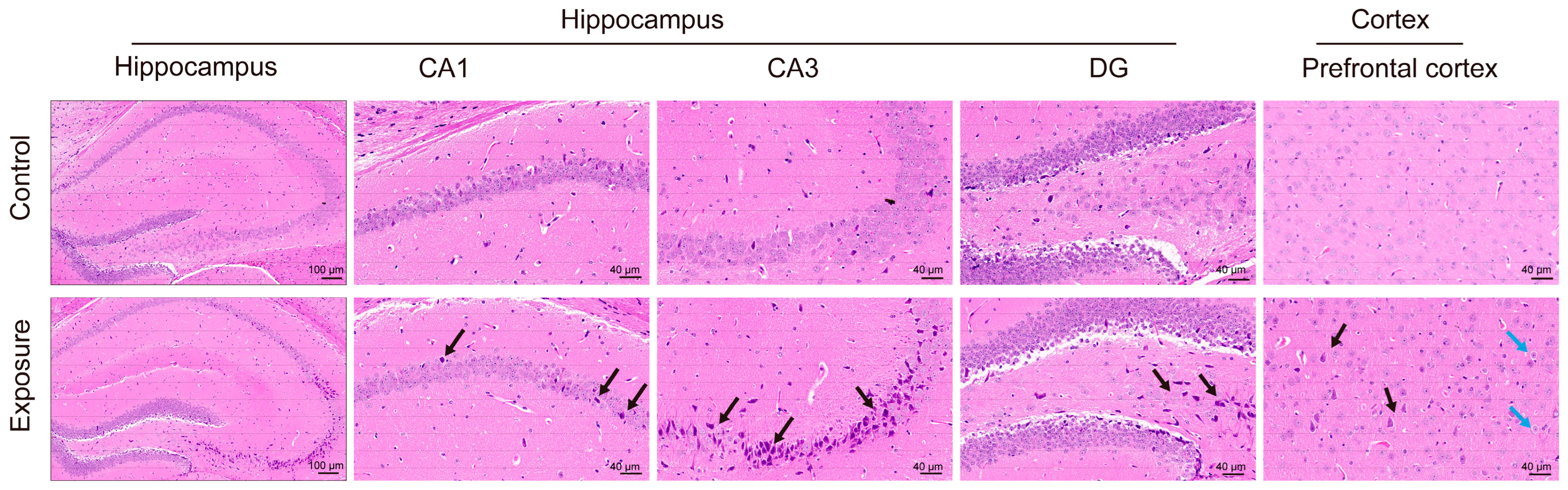
3.2. Effects of Air Pollution Exposure on the Hippocampus and Cortex
3.3. Alterations in the Gut Microbiome Induced by Air Pollution Exposure
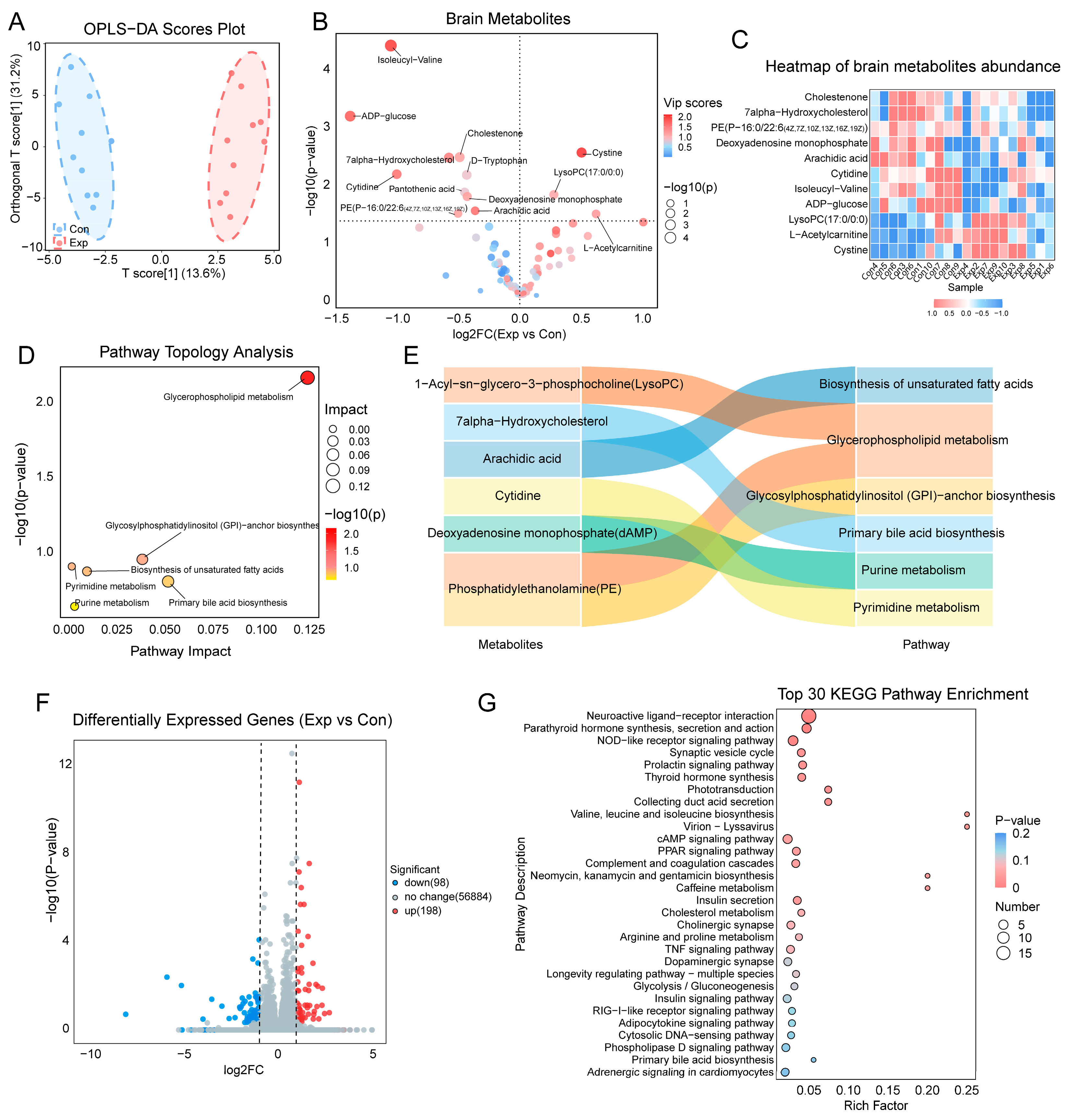
3.4. Metabolomic and Transcriptomic Alterations in the Intestine
3.5. Serum Metabolomic Alterations
3.6. Correlation Between the Gut Microbiome and Host Metabolome
3.7. Mediating Role of Intestinal and Serum Metabolites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Cohen, A.J.; Brauer, M.; Burnett, R.; Anderson, H.R.; Frostad, J.; Estep, K.; Balakrishnan, K.; Brunekreef, B.; Dandona, L.; Dandona, R.; et al. Estimates and 25-Year Trends of the Global Burden of Disease Attributable to Ambient Air Pollution: An Analysis of Data from the Global Burden of Diseases Study 2015. Lancet 2017, 389, 1907–1918. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.G.; Cole, T.B.; Dao, K.; Chang, Y.-C.; Coburn, J.; Garrick, J.M. Effects of Air Pollution on the Nervous System and Its Possible Role in Neurodevelopmental and Neurodegenerative Disorders. Pharmacol. Ther. 2020, 210, 107523. [Google Scholar] [CrossRef]
- Murata, H.; Barnhill, L.M.; Bronstein, J.M. Air Pollution and the Risk of Parkinson’s Disease: A Review. Mov. Disord. 2022, 37, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Castagna, A.; Mascheroni, E.; Fustinoni, S.; Montirosso, R. Air Pollution and Neurodevelopmental Skills in Preschool- and School-Aged Children: A Systematic Review. Neurosci. Biobehav. Rev. 2022, 136, 104623. [Google Scholar] [CrossRef]
- Chun, H.; Leung, C.; Wen, S.W.; McDonald, J.; Shin, H.H. Maternal Exposure to Air Pollution and Risk of Autism in Children: A Systematic Review and Meta-Analysis. Environ. Pollut. 2020, 256, 113307. [Google Scholar] [CrossRef]
- Pagalan, L.; Bickford, C.; Weikum, W.; Lanphear, B.; Brauer, M.; Lanphear, N.; Hanley, G.E.; Oberlander, T.F.; Winters, M. Association of Prenatal Exposure to Air Pollution with Autism Spectrum Disorder. JAMA Pediatr. 2019, 173, 86–92. [Google Scholar] [CrossRef]
- Morais, L.H.; Schreiber, H.L.; Mazmanian, S.K. The Gut Microbiota-Brain Axis in Behaviour and Brain Disorders. Nat. Rev. Microbiol. 2021, 19, 241–255. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, Q.; Liu, X. The Microbiota-Gut-Brain Axis and Neurodevelopmental Disorders. Protein Cell 2023, 14, 762–775. [Google Scholar] [CrossRef]
- Borre, Y.E.; O’Keeffe, G.W.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Microbiota and Neurodevelopmental Windows: Implications for Brain Disorders. Trends Mol. Med. 2014, 20, 509–518. [Google Scholar] [CrossRef]
- Desbonnet, L.; Clarke, G.; Traplin, A.; O’Sullivan, O.; Crispie, F.; Moloney, R.D.; Cotter, P.D.; Dinan, T.G.; Cryan, J.F. Gut Microbiota Depletion from Early Adolescence in Mice: Implications for Brain and Behaviour. Brain Behav. Immun. 2015, 48, 165–173. [Google Scholar] [CrossRef]
- Guilloteau, E.; Coll, P.; Lu, Z.; Djouina, M.; Cazaunau, M.; Waxin, C.; Bergé, A.; Caboche, S.; Gratien, A.; Al Marj, E.; et al. Murine in Utero Exposure to Simulated Complex Urban Air Pollution Disturbs Offspring Gut Maturation and Microbiota during Intestinal Suckling-to-Weaning Transition in a Sex-Dependent Manner. Part. Fibre Toxicol. 2022, 19, 41. [Google Scholar] [CrossRef]
- Xie, S.; Zhang, C.; Zhao, J.; Li, D.; Chen, J. Exposure to Concentrated Ambient PM2.5 (CAPM) Induces Intestinal Disturbance via Inflammation and Alternation of Gut Microbiome. Environ. Int. 2022, 161, 107138. [Google Scholar] [CrossRef]
- Li, T.; Fang, J.; Tang, S.; Du, H.; Zhao, L.; Wang, Y.; Deng, F.; Liu, Y.; Du, Y.; Cui, L.; et al. PM2.5 Exposure Associated with Microbiota Gut-Brain Axis: Multi-Omics Mechanistic Implications from the BAPE Study. Innovation 2022, 3, 100213. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Ji, Y.; Gao, H.; Pan, R.; Song, J.; He, Y.; Li, Y.; Wu, Y.; Yan, S.; Liang, Y.; et al. Effects of Urban Particulate Matter on Gut Microbiome and Partial Schizophrenia-like Symptoms in Mice: Evidence from Shotgun Metagenomic and Metabolomic Profiling. Sci. Total Environ. 2023, 857, 159305. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Xia, Y.; Wang, H.; Shi, L.; Yin, H.; Gu, M.; Yan, F. PM2.5 Induced Neurotoxicity through Unbalancing Vitamin B12 Metabolism by Gut Microbiota Disturbance. Gut Microbes 2023, 15, 2267186. [Google Scholar] [CrossRef] [PubMed]
- Dumas, M.-E.; Davidovic, L. Metabolic Profiling and Phenotyping of Central Nervous System Diseases: Metabolites Bring Insights into Brain Dysfunctions. J. Neuroimmune Pharmacol. 2015, 10, 402–424. [Google Scholar] [CrossRef]
- Cole, T.B.; Chang, Y.-C.; Dao, K.; Daza, R.; Hevner, R.; Costa, L.G. Developmental Exposure to Diesel Exhaust Upregulates Transcription Factor Expression, Decreases Hippocampal Neurogenesis, and Alters Cortical Lamina Organization: Relevance to Neurodevelopmental Disorders. J. Neurodev. Disord. 2020, 12, 41. [Google Scholar] [CrossRef]
- Nassan, F.L.; Wang, C.; Kelly, R.S.; Lasky-Su, J.A.; Vokonas, P.S.; Koutrakis, P.; Schwartz, J.D. Ambient PM2.5 Species and Ultrafine Particle Exposure and Their Differential Metabolomic Signatures. Environ. Int. 2021, 151, 106447. [Google Scholar] [CrossRef]
- Akyol, S.; Ugur, Z.; Yilmaz, A.; Ustun, I.; Gorti, S.K.K.; Oh, K.; McGuinness, B.; Passmore, P.; Kehoe, P.G.; Maddens, M.E.; et al. Lipid Profiling of Alzheimer’s Disease Brain Highlights Enrichment in Glycerol(Phospho)Lipid, and Sphingolipid Metabolism. Cells 2021, 10, 2591. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Q.; Su, Z.; Liu, Y.; Yan, T.; Zhang, Y.; Wang, T.; Wei, X.; Chen, Z.; Hu, G.; et al. Genetic Damage and Potential Mechanism Exploration under Different Air Pollution Patterns by Multi-Omics. Environ. Int. 2022, 170, 107636. [Google Scholar] [CrossRef]
- Xu, J.; Liu, Y.; Zhang, Q.; Su, Z.; Yan, T.; Zhou, S.; Wang, T.; Wei, X.; Chen, Z.; Hu, G.; et al. DNA Damage, Serum Metabolomic Alteration and Carcinogenic Risk Associated with Low-Level Air Pollution. Environ. Pollut. 2022, 297, 118763. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Wang, W.; Xia, J.; Jia, J.; Xu, J.; Dan, M.; Zhou, S.; Niu, P.; Gong, S.; Chen, Z.; et al. Exposure to the Real Ambient Air Pollutants Alters the Composition of Nasal Mucosa Bacteria in the Rat Model. Chemosphere 2022, 287, 132269. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.D. Prostaglandins and Leukotrienes: Advances in Eicosanoid Biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef]
- Guxens, M.; Garcia-Esteban, R.; Giorgis-Allemand, L.; Forns, J.; Badaloni, C.; Ballester, F.; Beelen, R.; Cesaroni, G.; Chatzi, L.; de Agostini, M.; et al. Air Pollution during Pregnancy and Childhood Cognitive and Psychomotor Development: Six European Birth Cohorts. Epidemiology 2014, 25, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Holm, S.M.; Balmes, J.R.; Gunier, R.B.; Kogut, K.; Harley, K.G.; Eskenazi, B. Cognitive Development and Prenatal Air Pollution Exposure in the CHAMACOS Cohort. Environ. Health Perspect. 2023, 131, 37007. [Google Scholar] [CrossRef]
- Ni, Y.; Loftus, C.T.; Szpiro, A.A.; Young, M.T.; Hazlehurst, M.F.; Murphy, L.E.; Tylavsky, F.A.; Mason, W.A.; LeWinn, K.Z.; Sathyanarayana, S.; et al. Associations of Pre- and Postnatal Air Pollution Exposures with Child Behavioral Problems and Cognitive Performance: A U.S. Multi-Cohort Study. Environ. Health Perspect. 2022, 130, 67008. [Google Scholar] [CrossRef]
- Zhang, T.; Zheng, X.; Wang, X.; Zhao, H.; Wang, T.; Zhang, H.; Li, W.; Shen, H.; Yu, L. Maternal Exposure to PM2.5 during Pregnancy Induces Impaired Development of Cerebral Cortex in Mice Offspring. Int. J. Mol. Sci. 2018, 19, 257. [Google Scholar] [CrossRef]
- Allen, J.L.; Liu, X.; Pelkowski, S.; Palmer, B.; Conrad, K.; Oberdörster, G.; Weston, D.; Mayer-Pröschel, M.; Cory-Slechta, D.A. Early Postnatal Exposure to Ultrafine Particulate Matter Air Pollution: Persistent Ventriculomegaly, Neurochemical Disruption, and Glial Activation Preferentially in Male Mice. Environ. Health Perspect. 2014, 122, 939–945. [Google Scholar] [CrossRef]
- Liu, F.; Liu, C.; Liu, Y.; Wang, J.; Wang, Y.; Yan, B. Neurotoxicity of the Air-Borne Particles: From Molecular Events to Human Diseases. J. Hazard. Mater. 2023, 457, 131827. [Google Scholar] [CrossRef]
- Fonken, L.K.; Xu, X.; Weil, Z.M.; Chen, G.; Sun, Q.; Rajagopalan, S.; Nelson, R.J. Air Pollution Impairs Cognition, Provokes Depressive-like Behaviors and Alters Hippocampal Cytokine Expression and Morphology. Mol. Psychiatry 2011, 16, 987–995. [Google Scholar] [CrossRef]
- Mutlu, E.A.; Comba, I.Y.; Cho, T.; Engen, P.A.; Yazıcı, C.; Soberanes, S.; Hamanaka, R.B.; Niğdelioğlu, R.; Meliton, A.Y.; Ghio, A.J.; et al. Inhalational Exposure to Particulate Matter Air Pollution Alters the Composition of the Gut Microbiome. Environ. Pollut. 2018, 240, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, J.; Chen, M.; Huang, X.; Xie, X.; Li, W.; Cao, Q.; Kan, H.; Xu, Y.; Ying, Z. Exposure to Concentrated Ambient PM2.5 Alters the Composition of Gut Microbiota in a Murine Model. Part. Fibre Toxicol. 2018, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, T.; Si, B.; Du, H.; Liu, Y.; Waqas, A.; Huang, S.; Zhao, G.; Chen, S.; Xu, A. Intratracheally Instillated Diesel PM2.5 Significantly Altered the Structure and Composition of Indigenous Murine Gut Microbiota. Ecotoxicol. Environ. Saf. 2021, 210, 111903. [Google Scholar] [CrossRef] [PubMed]
- Naseribafrouei, A.; Hestad, K.; Avershina, E.; Sekelja, M.; Linløkken, A.; Wilson, R.; Rudi, K. Correlation between the Human Fecal Microbiota and Depression. Neurogastroenterol. Motil. 2014, 26, 1155–1162. [Google Scholar] [CrossRef]
- Parker, B.J.; Wearsch, P.A.; Veloo, A.C.M.; Rodriguez-Palacios, A. The Genus Alistipes: Gut Bacteria with Emerging Implications to Inflammation, Cancer, and Mental Health. Front. Immunol. 2020, 11, 906. [Google Scholar] [CrossRef]
- Li, R.; Wang, Y.; Hou, B.; Lam, S.M.; Zhang, W.; Chen, R.; Shui, G.; Sun, Q.; Qiang, G.; Liu, C. Lipidomics Insight into Chronic Exposure to Ambient Air Pollution in Mice. Environ. Pollut. 2020, 262, 114668. [Google Scholar] [CrossRef]
- Guido, M.E.; Monjes, N.M.; Wagner, P.M.; Salvador, G.A. Circadian Regulation and Clock-Controlled Mechanisms of Glycerophospholipid Metabolism from Neuronal Cells and Tissues to Fibroblasts. Mol. Neurobiol. 2022, 59, 326–353. [Google Scholar] [CrossRef]
- Mochel, F. Lipids and Synaptic Functions. J. Inherit. Metab. Dis. 2018, 41, 1117–1122. [Google Scholar] [CrossRef]
- Qian, X.; Hai, W.; Chen, S.; Zhang, M.; Jiang, X.; Tang, H. Multi-Omics Data Reveals Aberrant Gut Microbiota-Host Glycerophospholipid Metabolism in Association with Neuroinflammation in APP/PS1 Mice. Gut Microbes 2023, 15, 2282790. [Google Scholar] [CrossRef]
- Zheng, P.; Wu, J.; Zhang, H.; Perry, S.W.; Yin, B.; Tan, X.; Chai, T.; Liang, W.; Huang, Y.; Li, Y.; et al. The Gut Microbiome Modulates Gut-Brain Axis Glycerophospholipid Metabolism in a Region-Specific Manner in a Nonhuman Primate Model of Depression. Mol. Psychiatry 2021, 26, 2380–2392. [Google Scholar] [CrossRef]
- Tian, T.; Mao, Q.; Xie, J.; Wang, Y.; Shao, W.-H.; Zhong, Q.; Chen, J.-J. Multi-Omics Data Reveals the Disturbance of Glycerophospholipid Metabolism Caused by Disordered Gut Microbiota in Depressed Mice. J. Adv. Res. 2022, 39, 135–145. [Google Scholar] [CrossRef]
- Xie, J.; Zhong, Q.; Wu, W.-T.; Chen, J.-J. Multi-Omics Data Reveals the Important Role of Glycerophospholipid Metabolism in the Crosstalk between Gut and Brain in Depression. J. Transl. Med. 2023, 21, 93. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-Y.; Huang, B.X.; Spector, A.A. Molecular and Signaling Mechanisms for Docosahexaenoic Acid-Derived Neurodevelopment and Neuroprotection. Int. J. Mol. Sci. 2022, 23, 4635. [Google Scholar] [CrossRef]
- Ferreira, I.; Falcato, F.; Bandarra, N.; Rauter, A.P. Resolvins, Protectins, and Maresins: DHA-Derived Specialized pro-Resolving Mediators, Biosynthetic Pathways, Synthetic Approaches, and Their Role in Inflammation. Molecules 2022, 27, 1677. [Google Scholar] [CrossRef]
- Sergeant, S.; Rahbar, E.; Chilton, F.H. Gamma-Linolenic Acid, Dihommo-Gamma Linolenic, Eicosanoids and Inflammatory Processes. Eur. J. Pharmacol. 2016, 785, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.Y.; Song, J. Important Roles of Linoleic Acid and α-Linolenic Acid in Regulating Cognitive Impairment and Neuropsychiatric Issues in Metabolic-Related Dementia. Life Sci. 2024, 337, 122356. [Google Scholar] [CrossRef] [PubMed]
- Dave, A.; Park, E.-J.; Kumar, A.; Parande, F.; Beyoğlu, D.; Idle, J.R.; Pezzuto, J.M. Consumption of Grapes Modulates Gene Expression, Reduces Non-Alcoholic Fatty Liver Disease, and Extends Longevity in Female C57BL/6J Mice Provided with a High-Fat Western-Pattern Diet. Foods 2022, 11, 1984. [Google Scholar] [CrossRef]
- Thierer, J.H.; Foresti, O.; Yadav, P.K.; Wilson, M.H.; Moll, T.O.C.; Shen, M.-C.; Busch-Nentwich, E.M.; Morash, M.; Mohlke, K.L.; Rawls, J.F.; et al. Pla2g12b Drives Expansion of Triglyceride-Rich Lipoproteins. Nat. Commun. 2024, 15, 2095. [Google Scholar] [CrossRef]
- Xu, J.; Ma, C.; Hua, M.; Li, J.; Xiang, Z.; Wu, J. CNS and CNS Diseases in Relation to Their Immune System. Front. Immunol. 2022, 13, 1063928. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Omega-3 Fatty Acids in Inflammation and Autoimmune Diseases. J. Am. Coll. Nutr. 2002, 21, 495–505. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Control (Mean ± SD) | Exposure (Mean ± SD) | t | p |
|---|---|---|---|---|
| Shannon index | 6.50 ± 0.42 | 6.62 ± 0.27 | −0.73 | 0.479 |
| Chao1 | 33,650.24 ± 18,032.92 | 34,495.96 ± 16,589.99 | −0.11 | 0.914 |
| Observed species | 8378.50 ± 696.35 | 8805.20 ± 579.11 | −1.49 | 0.154 |
| Simpson index | 0.97 ± 0.01 | 0.96 ± 0.01 | 0.32 | 0.754 |
| ACE index | 9255.11 ± 890.40 | 9704.75 ± 569.13 | −1.35 | 0.198 |
| Outcome Indicator | Exposure | Mediators | Indirect Effect (95% CI) | Proportion Mediated (%) |
|---|---|---|---|---|
| brain_LysoPC(17:0/0:0) | s__Muribaculaceae_bacterium_Isolate_037_Harlan_ | intestinal_Cer(d18:1/16:0) | 0.493 (0.106, 1.136) | 75.4 |
| s__Muribaculaceae_bacterium_Isolate_037_Harlan_ | serum_Esterase | 0.377 (0.083, 0.792) | 57.8 | |
| s__Muribaculaceae_bacterium_Isolate_037_Harlan_ | serum_PS(18:1(11Z)/20:0) | 0.332 (0.015, 0.882) | 50.8 | |
| s__Bacteroidales_bacterium | intestinal_Cer(d18:1/16:0) | 0.485 (0.073, 1.11) | 77.3 | |
| s__Bacteroidales_bacterium | serum_Esterase | 0.368 (0.076, 0.763) | 58.7 | |
| s__Bacteroidales_bacterium | serum_PS(18:1(11Z)/20:0) | 0.343 (0.068, 0.833) | 54.6 | |
| s_Bacteroidales_bacterium | serum_gamma-Linolenic acid | 0.283 (0.011, 0.736) | 45.2 | |
| o__Coriobacteriales | serum_PS(18:1(11Z)/20:0) | −0.102 (−0.23, −0.021) | 46.9 | |
| f__Coriobacteriaceae | serum_PS(18:1(11Z)/20:0) | −0.104 (−0.242, −0.022) | 46.6 | |
| s__Bacteroidales_bacterium_55_9 | serum_Esterase | 0.112 (0.004, 0.275) | 90.1 | |
| s__Bacteroidales_bacterium_55_9 | serum_16-Hydroxyhexadecanoic acid | 0.041 (0.003, 0.172) | 32.7 | |
| s__Coriobacteriaceae_bacterium | serum_PS(18:1(11Z)/20:0) | −0.064 (−0.189, −0.011) | 72.7 | |
| s__Alistipes_sp_58_9_plus | serum_Dimyristoylphosphatidylcholine | 0.074 (0.004, 0.148) | 66.2 | |
| s__Alistipes_sp_ | serum_Leukotriene C4 | 0.097 (0.007, 0.213) | 78.8 | |
| s__Bacteroides_caecimuris | intestinal_Cer(d18:1/16:0) | 0.271 (0.042, 0.554) | 98.4 | |
| s__Bacteroides_caecimuris | serum_Esterase | 0.228 (0.045, 0.576) | 83 | |
| s__Bacteroides_caecimuris | intestinal_Ubiquinone-2 | 0.164 (0.017, 0.395) | 59.7 | |
| s__Bacteroides_caecimuris | serum_PS(18:1(11Z)/20:0) | 0.216 (0.064, 0.477) | 78.4 | |
| s__Bacteroides_caecimuris | serum_Leukotriene C4 | 0.132 (0.01, 0.326) | 48 | |
| s__Bacteroides_caecimuris | serum_gamma-Linolenic acid | 0.134 (0.006, 0.415) | 48.9 | |
| brain_PE(P−16:0/22:6(4Z,7Z,10Z,13Z,16Z,19Z)) | f__Rikenellaceae | serum_Arachidoyl Ethanolamide | −0.138 (−0.322, −0.014) | 87.6 |
| f__Rikenellaceae | serum_Heptadecanoic acid | −0.142 (−0.365, −0.018) | 89.8 | |
| f__Rikenellaceae | serum_Palmitoylethanolamide | −0.133 (−0.309, −0.028) | 84.1 | |
| g_Alistipes | serum_Arachidoyl Ethanolamide | −0.128 (−0.288, −0.003) | 94.3 | |
| g_Alistipes | serum_Palmitoylethanolamide | −0.122 (−0.288, −0.01) | 90.2 | |
| g__Duncaniella | serum_Phenyllactic acid | 0.207 (0.032, 0.48) | 51.6 | |
| g__Duncaniella | serum_Sphingosine | 0.397 (0.002, 1.091) | 98.7 | |
| g_Heminiphilus | serum_Phenyllactic acid | −0.11 (−0.237, −0.01) | 52.3 | |
| s__Duncaniella_dubosii | serum_Phenyllactic acid | 0.114 (0.008, 0.248) | 37.1 | |
| g_Allobaculum | serum_Phenyllactic acid | 0.032 (0.005, 0.061) | 55.4 | |
| s_Allobaculum_sp_539 | serum_Phenyllactic acid | 0.032 (0.01, 0.059) | 74.1 | |
| s__Alistipes_senegalensis | serum_Arachidoyl Ethanolamide | −0.113 (−0.256, −0.007) | 72.9 | |
| s_Alistipes_finegoldii | serum_Arachidoyl Ethanolamide | −0.099 (−0.231, −0.003) | 69.5 | |
| s_Alistipes_finegoldii | serum_Heptadecanoic acid | −0.101 (−0.226, −0.002) | 70.9 | |
| s__Alistipes_onderdonkii | serum_Arachidoyl Ethanolamide | −0.1 (−0.257, −0.02) | 74 | |
| s__Alistipes_onderdonkii | serum_Heptadecanoic acid | −0.103 (−0.238, −0.001) | 75.8 | |
| s__Alistipes_shahii | serum_Heptadecanoic acid | −0.147 (−0.329, −0.022) | 86.4 | |
| s__Alistipes_timonensis | serum_Palmitoylethanolamide | −0.096 (−0.223, −0.007) | 63.7 | |
| s__Alistipes_sp_An66 | serum_Arachidoyl Ethanolamide | −0.1 (−0.24, −0.003) | 78.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Zhang, Y.; Ran, S.; Zhang, J.; Tian, F.; Shi, H.; Wei, S.; Li, X.; Li, X.; Gao, Y.; et al. A Multi-Omics Study of Neurodamage Induced by Growth-Stage Real-Time Air Pollution Exposure in Mice via the Microbiome–Gut–Brain Axis. Toxics 2025, 13, 260. https://doi.org/10.3390/toxics13040260
Yang Z, Zhang Y, Ran S, Zhang J, Tian F, Shi H, Wei S, Li X, Li X, Gao Y, et al. A Multi-Omics Study of Neurodamage Induced by Growth-Stage Real-Time Air Pollution Exposure in Mice via the Microbiome–Gut–Brain Axis. Toxics. 2025; 13(4):260. https://doi.org/10.3390/toxics13040260
Chicago/Turabian StyleYang, Zijun, Yi Zhang, Shanshan Ran, Jingyi Zhang, Fei Tian, Hui Shi, Shengtao Wei, Xiuxiu Li, Xinyue Li, Yonggui Gao, and et al. 2025. "A Multi-Omics Study of Neurodamage Induced by Growth-Stage Real-Time Air Pollution Exposure in Mice via the Microbiome–Gut–Brain Axis" Toxics 13, no. 4: 260. https://doi.org/10.3390/toxics13040260
APA StyleYang, Z., Zhang, Y., Ran, S., Zhang, J., Tian, F., Shi, H., Wei, S., Li, X., Li, X., Gao, Y., Jia, G., Lin, H., Chen, Z., & Zhang, Z. (2025). A Multi-Omics Study of Neurodamage Induced by Growth-Stage Real-Time Air Pollution Exposure in Mice via the Microbiome–Gut–Brain Axis. Toxics, 13(4), 260. https://doi.org/10.3390/toxics13040260