
Updates on Oxaliplatin-Induced Peripheral Neurotoxicity (OXAIPN)


{kind=link}
Abstract
:1. Introduction
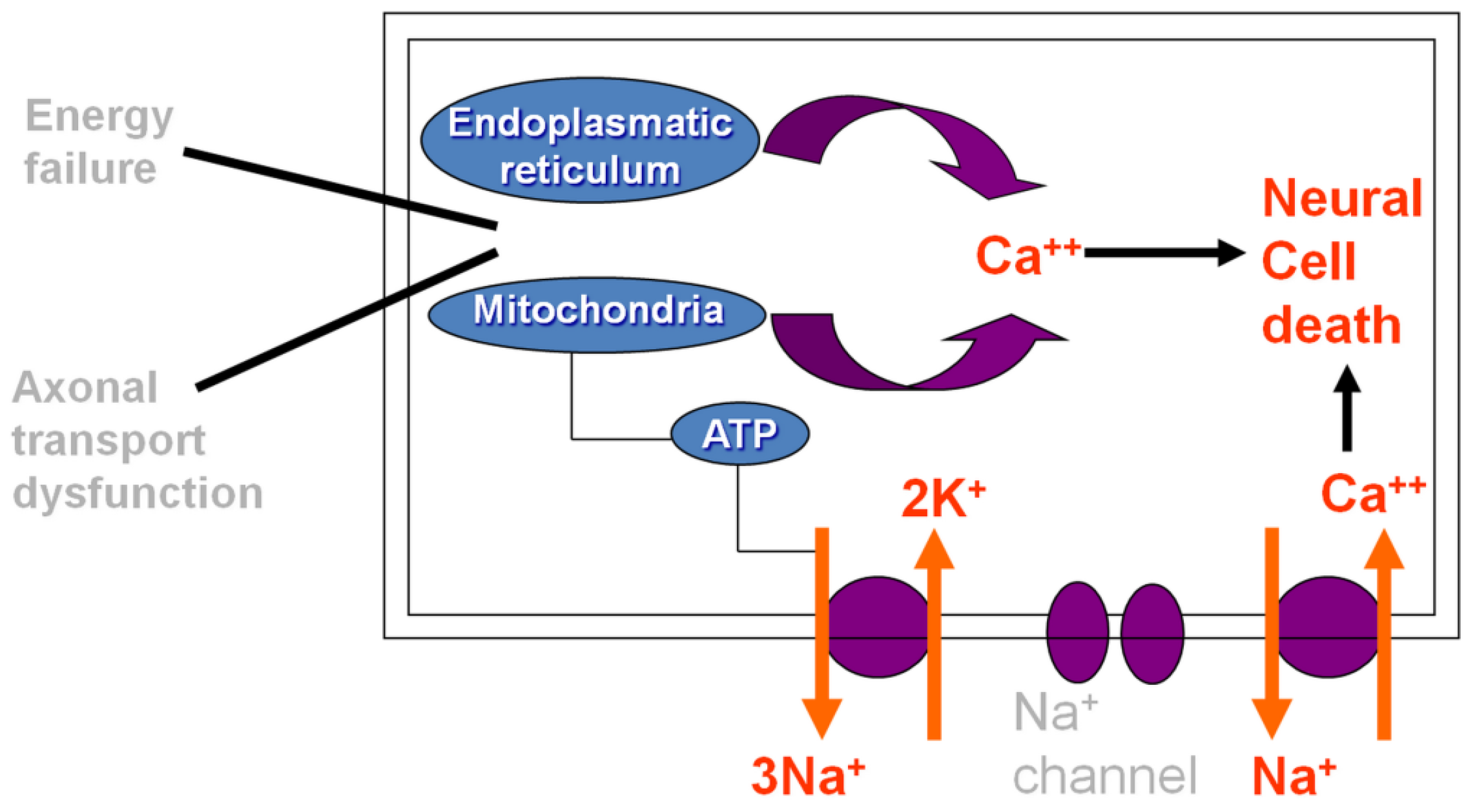
2. Pathogenesis

3. Incidence, Severity and Risk Factors
4. Clinical and Electrophysiological Characteristics
5. Long Term Outcome
6. Pharmacogenetics
7. Treatment Options
8. Conclusions
Conflicts of Interest
References
- Kim, G.P.; Erlichman, C. Oxaliplatin in the treatment of colorectal cancer. Expert Opin. Drug Metab. Toxicol. 2007, 3, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Bruna, J.; Marmiroli, P.; Cavaletti, G. Chemotherapy-induced peripheral neurotoxicity (CIPN): An update. Crit. Rev. Oncol. Hematol. 2012, 82, 51–77. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Zolota, V.; Kyriakopoulou, O.; Kalofonos, H.P. Toxic peripheral neuropathy associated with commonly used chemotherapeutic agents. J. BUON 2010, 15, 435–446. [Google Scholar] [PubMed]
- Cavaletti, G.; Marmiroli, P. Chemotherapy-induced peripheral neurotoxicity. Nat. Rev. Neurol. 2010, 6, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Polychronopoulos, P.; Koutras, A.; Xiros, N.; Petsas, T.; Argyriou, K.; Chroni, E.; Kalofonos, H.P. Clinical and electrophysiological features of peripheral neuropathy induced by administration of cisplatin plus paclitaxel-based chemotherapy. Eur. J. Cancer Care Engl. 2007, 16, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Park, S.B.; Krishnan, A.V.; Lin, C.S.; Goldstein, D.; Friedlander, M.; Kiernan, M.C. Mechanisms underlying chemotherapy-induced neurotoxicity and the potential for neuroprotective strategies. Curr. Med. Chem. 2008, 15, 3081–3094. [Google Scholar] [CrossRef] [PubMed]
- Carozzi, V.A.; Canta, A.; Chiorazzi, A.; Cavaletti, G. Chemotherapy-induced peripheral neuropathy: What do we know about mechanisms? Neurosci. Lett. 2014. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Polychronopoulos, P.; Iconomou, G.; Chroni, E.; Kalofonos, H.P. A review on oxaliplatin-induced peripheral nerve damage. Cancer Treat. Rev. 2008, 34, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare Mannelli, L.; Zanardelli, M.; Failli, P.; Ghelardini, C. Oxaliplatin-induced neuropathy: Oxidative stress as pathological mechanism. Protective effect of silibinin. J. Pain 2012, 13, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Renn, C.L.; Carozzi, V.A.; Rhee, P.; Gallop, D.; Dorsey, S.G.; Cavaletti, G. Multimodal assessment of painful peripheral neuropathy induced by chronic oxaliplatin-based chemotherapy in mice. Mol. Pain 2011, 26, 29. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare Mannelli, L.; Pacini, A.; Bonaccini, L.; Zanardelli, M.; Mello, T.; Ghelardini, C. Morphologic features and glial activation in rat oxaliplatin-dependent neuropathic pain. J. Pain. 2014, 14, 1585–1600. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare Mannelli, L.; Pacini, A.; Micheli, L.; Tani, A.; Zanardelli, M.; Ghelardini, C. Glial role in oxaliplatin-induced neuropathic pain. Exp. Neurol. 2014, 261, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Kyritsis, A.P.; Makatsoris, T.; Kalofonos, H.P. Chemotherapy-induced peripheral neuropathy in adults: A comprehensive update of the literature. Cancer Manag. Res. 2014, 6, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Cavaletti, G.; Briani, C.; Velasco, R.; Bruna, J.; Campagnolo, M.; Alberti, P.; Bergamo, F.; Cortinovis, D.; Cazzaniga, M.; et al. Clinical pattern and associations of oxaliplatin acute neurotoxicity: A prospective study in 170 patients with colorectal cancer. Cancer 2013, 119, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Park, S.B.; Lin, C.S.; Krishnan, A.V.; Goldstein, D.; Friedlander, M.L.; Kiernan, M.C. Oxaliplatin-induced neurotoxicity: Changes in axonal excitability precede development of neuropathy. Brain 2009, 132, 2712–2723. [Google Scholar] [CrossRef] [PubMed]
- De Gramont, A.; Figer, A.; Seymour, M.; Homerin, M.; Hmissi, A.; Cassidy, J.; Cortes-Funes, H.; Boni, C.; Cervantes, A.; Freyer, G.; et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J. Clin. Oncol. 2000, 18, 2938–2947. [Google Scholar] [PubMed]
- Kemeny, N.; Garay, C.A.; Gurtler, J.; Hochster, H.; Kennedy, P.; Benson, A.; Brandt, D.S.; Polikoff, J.; Wertheim, M.; Shumaker, G.; et al. Randomized multicenter phase II trial of bolus plus infusional fluorouracil/leucovorin compared with fluorouracil/leucovorin plus oxaliplatin as third-line treatment of patients with advanced colorectal cancer. J. Clin. Oncol. 2004, 22, 4753–4761. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Cavaletti, G.; Antonacopoulou, A.; Genazzani, A.A.; Briani, C.; Bruna, J.; Terrazzino, S.; Velasco, R.; Alberti, P.; Campagnolo, M.; et al. Voltage-gated sodium channel polymorphisms play a pivotal role in the development of oxaliplatin-induced peripheral neurotoxicity: Results from a prospective multicenter study. Cancer 2013, 119, 3570–3577. [Google Scholar] [CrossRef] [PubMed]
- Velasco, R.; Bruna, J.; Briani, C.; Argyriou, A.A.; Cavaletti, G.; Alberti, P.; Cacciavillani, M.; Frigeni, B.; Lonardi, S.; Cortinovis, D.; et al. Early predictors of oxaliplatin-induced cumulative neuropathy in colorectal cancer patients. J. Neurol. Neurosurg. Psychiatry 2014, 85, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Sioka, C.; Kyritsis, A.P. Central and peripheral nervous system toxicity of common chemotherapeutic agents. Cancer Chemother. Pharmacol. 2009, 63, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Storey, D.J.; Sakala, M.; McLean, C.M.; Phillips, H.A.; Dawson, L.K.; Wall, L.R.; Fallon, M.T.; Clive, S. Capecitabine combined with oxaliplatin (CapOx) in clinical practice: How significant is peripheral neuropathy? Ann. Oncol. 2010, 21, 1657–1661. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Velasco, R.; Briani, C.; Cavaletti, G.; Bruna, J.; Alberti, P.; Cacciavillani, M.; Lonardi, S.; Santos, C.; Cortinovis, D.; et al. Peripheral neurotoxicity of oxaliplatin in combination with 5-fluorouracil (FOLFOX) or capecitabine (XELOX): A prospective evaluation of 150 colorectal cancer patients. Ann. Oncol. 2012, 23, 3116–3122. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, R.M.; Tabah-Fisch, I.; Bleiberg, H.; de Gramont, A.; Tournigand, C.; Andre, T.; Rothenberg, M.L.; Green, E.; Sargent, D.J. Pooled analysis of safety and efficacy of oxaliplatin plus fluorouracil/leucovorin administered bimonthly in elderly patients with colorectal cancer. J. Clin. Oncol. 2006, 24, 4085–4091. [Google Scholar] [CrossRef] [PubMed]
- Baek, K.K.; Lee, J.; Park, S.H.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Cho, Y.B.; Yun, S.H.; Kim, H.C.; et al. Oxaliplatin-induced chronic peripheral neurotoxicity: A prospective analysis in patients with colorectal cancer. Cancer Res. Treat. 2010, 42, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Briani, C.; Cavaletti, G.; Bruna, J.; Alberti, P.; Velasco, R.; Lonardi, S.; Cortinovis, D.; Cazzaniga, M.; Campagnolo, M.; et al. Advanced age and liability to oxaliplatin-induced peripheral neuropathy: Post hoc analysis of a prospective study. Eur. J. Neurol. 2013, 20, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Avan, A.; Postma, T.J.; Ceresa, C.; Avan, A.; Cavaletti, G.; Giovannetti, E.; Peters, G.J. Platinum-Induced Neurotoxicity and Preventive Strategies: Past, Present, and Future. Oncologist 2015, 20, 411–432. [Google Scholar] [CrossRef] [PubMed]
- Lucchetta, M.; Lonardi, S.; Bergamo, F.; Alberti, P.; Velasco, R.; Argyriou, A.A.; Briani, C.; Bruna, J.; Cazzaniga, M.; Cortinovis, D.; et al. Incidence of atypical acute nerve hyperexcitability symptoms in oxaliplatin-treated patients with colorectal cancer. Cancer Chemother. Pharmacol. 2012, 70, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.H.; Lehky, T.; Thomas, R.R.; Quinn, M.G.; Floeter, M.K.; Grem, J.L. Acute oxaliplatin-induced peripheral nerve hyperexcitability. J. Clin. Oncol. 2002, 20, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Polychronopoulos, P.; Iconomou, G.; Koutras, A.; Makatsoris, T.; Gerolymos, M.K.; Gourzis, P.; Assimakopoulos, K.; Kalofonos, H.P.; Chroni, E. Incidence and characteristics of peripheral neuropathy during oxaliplatin-based chemotherapy for metastatic colon cancer. Acta Oncol. 2007, 46, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Griffith, K.A.; Dorsey, S.G.; Renn, C.L.; Zhu, S.; Johantgen, M.E.; Cornblath, D.R.; Argyriou, A.A.; Cavaletti, G.; Merkies, I.S.; Alberti, P.; et al. Correspondence between neurophysiological and clinical measurements of chemotherapy-induced peripheral neuropathy: Secondary analysis of data from the CI-PeriNomS study. J. Peripher. Nerv. Syst. 2014, 19, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Briani, C.; Argyriou, A.A.; Izquierdo, C.; Velasco, R.; Campagnolo, M.; Alberti, P.; Frigeni, B.; Cacciavillani, M.; Bergamo, F.; Cortinovis, D.; et al. Long-term course of oxaliplatin-induced polyneuropathy: A prospective 2-year follow-up study. J. Peripher. Nerv. Syst. 2014, 19, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Vatandoust, S.; Joshi, R.; Pittman, K.B.; Esterman, A.; Broadbridge, V.; Adams, J.; Singhal, N.; Yeend, S.; Price, T.J. A descriptive study of persistent oxaliplatin-induced peripheral neuropathy in patients with colorectal cancer. Support. Care Cancer 2014, 22, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Townsend, A.R.; Bishnoi, S.; Broadbridge, V.; Beeke, C.; Karapetis, C.S.; Jain, K.; Luke, C.; Padbury, R.; Price, T.J. Rechallenge with oxaliplatin and fluoropyrimidine for metastatic colorectal carcinoma after prior therapy. Am. J. Clin. Oncol. 2013, 36, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Cavaletti, G.; Alberti, P.; Marmiroli, P. Chemotherapy-induced peripheral neurotoxicity in the era of pharmacogenomics. Lancet Oncol. 2011, 12, 1551–1561. [Google Scholar] [CrossRef]
- Cecchin, E.; D’Andrea, M.; Lonardi, S.; Zanusso, C.; Pella, N.; Errante, D.; de Mattia, E.; Polesel, J.; Innocenti, F.; Toffoli, G. A prospective validation pharmacogenomic study in the adjuvant setting of colorectal cancer patients treated with the 5-fluorouracil/leucovorin/oxaliplatin (FOLFOX4) regimen. Pharmacogenomics J. 2013, 13, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Antonacopoulou, A.G.; Argyriou, A.A.; Scopa, C.D.; Kottorou, A.; Peroukides, S.; Kalofonos, H.P.; Kominea, A. Integrin beta-3 L33P: A new insight into the pathogenesis of chronic oxaliplatin-induced peripheral neuropathy? Eur. J. Neurol. 2010, 17, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Antonacopoulou, A.G.; Scopa, C.D.; Kottorou, A.; Kominea, A.; Peroukides, S.; Kalofonos, H.P. Liability of the voltage-gated sodium channel gene SCN2A R19K polymorphism to oxaliplatin-induced peripheral neuropathy. Oncology 2009, 77, 254–256. [Google Scholar] [CrossRef] [PubMed]
- Won, H.H.; Lee, J.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Kim, J.W.; Lee, S.Y.; Park, S.H. Polymorphic markers associated with severe oxaliplatin-induced, chronic peripheral neuropathy in colon cancer patients. Cancer 2012, 118, 2828–2836. [Google Scholar] [CrossRef] [PubMed]
- Oguri, T.; Mitsuma, A.; Inada-Inoue, M.; Morita, S.; Shibata, T.; Shimokata, T.; Sugishita, M.; Nakayama, G.; Uehara, K.; Hasegawa, Y.; et al. Genetic polymorphisms associated with oxaliplatin-induced peripheral neurotoxicity in Japanese patients with colorectal cancer. Int. J. Clin. Pharmacol. Ther. 2013, 51, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Terrazzino, S.; Argyriou, A.A.; Cargnin, S.; Antonacopoulou, A.G.; Briani, C.; Bruna, J.; Velasco, R.; Alberti, P.; Campagnolo, M.; Lonardi, S.; et al. Genetic determinants of chronic oxaliplatin-induced peripheral neurotoxicity: A genome-wide study replication and meta-analysis. J. Peripher. Nerv. Syst. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gamelin, L.; Boisdron-Celle, M.; Delva, R.; Guérin-Meyer, V.; Ifrah, N.; Morel, A.; Gamelin, E. Prevention of oxaliplatin-related neurotoxicity by calcium and magnesium infusions: A retrospective study of 161 patients receiving oxaliplatin combined with 5-Fluorouracil and leucovorin for advanced colorectal cancer. Clin. Cancer Res. 2004, 10, 4055–4061. [Google Scholar] [CrossRef] [PubMed]
- Hochster, H.S.; Grothey, A.; Childs, B.H. Use of calcium and magnesium salts to reduce oxaliplatin-related neurotoxicity. J. Clin. Oncol. 2007, 25, 4028–4029. [Google Scholar] [CrossRef] [PubMed]
- Gamelin, L.; Boisdron-Celle, M.; Morel, A.; Poirier, A.L.; Berger, V.; Gamelin, E.; Tournigand, C.; de Gramont, A. Oxaliplatin-related neurotoxicity: Interest of calcium-magnesium infusion and no impact on its efficacy. J. Clin. Oncol. 2008, 26, 1188–1189, Author reply 1189–1190. [Google Scholar] [CrossRef] [PubMed]
- Loprinzi, C.L.; Qin, R.; Dakhil, S.R.; Fehrenbacher, L.; Flynn, K.A.; Atherton, P.; Seisler, D.; Qamar, R.; Lewis, G.C.; Grothey, A. Phase III randomized, placebo-controlled, double-blind study of intravenous calcium and magnesium to prevent oxaliplatin-induced sensory neurotoxicity (N08CB/Alliance). J. Clin. Oncol. 2014, 32, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Albers, J.W.; Chaudhry, V.; Cavaletti, G.; Donehower, R.C. Interventions for preventing neuropathy caused by cisplatin and related compounds. Cochrane Database Syst. Rev. 2011, 2, CD005228. [Google Scholar] [PubMed]
- Tournigand, C.; Cervantes, A.; Figer, A.; Lledo, G.; Flesch, M.; Buyse, M.; Mineur, L.; Carola, E.; Etienne, P.L.; Rivera, F.; et al. OPTIMOX1: A randomized study of FOLFOX4 or FOLFOX7 with oxaliplatin in a stop-and-Go fashion in advanced colorectal cancer-a GERCOR study. J. Clin. Oncol. 2006, 24, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.M.; Pang, H.; Cirrincione, C.; Fleishman, S.; Paskett, E.D.; Ahles, T.; Bressler, L.R.; Fadul, C.E.; Knox, C.; Le-Lindqwister, N.; et al. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: A randomized clinical trial. JAMA 2013, 309, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Sereno, M.; Gutiérrez-Gutiérrez, G.; Gómez-Raposo, C.; López-Gómez, M.; Merino-Salvador, M.; Tébar, F.Z.; Rodriguez-Antona, C.; Casado, E. Oxaliplatin induced-neuropathy in digestive tumors. Crit. Rev. Oncol. Hematol. 2014, 89, 166–178. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Argyriou, A.A. Updates on Oxaliplatin-Induced Peripheral Neurotoxicity (OXAIPN). Toxics 2015, 3, 187-197. https://doi.org/10.3390/toxics3020187
Argyriou AA. Updates on Oxaliplatin-Induced Peripheral Neurotoxicity (OXAIPN). Toxics. 2015; 3(2):187-197. https://doi.org/10.3390/toxics3020187
Chicago/Turabian StyleArgyriou, Andreas A. 2015. "Updates on Oxaliplatin-Induced Peripheral Neurotoxicity (OXAIPN)" Toxics 3, no. 2: 187-197. https://doi.org/10.3390/toxics3020187
APA StyleArgyriou, A. A. (2015). Updates on Oxaliplatin-Induced Peripheral Neurotoxicity (OXAIPN). Toxics, 3(2), 187-197. https://doi.org/10.3390/toxics3020187