Radiocarbon Tracers in Toxicology and Medicine: Recent Advances in Technology and Science

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Technology
2.1. Graphite
2.2. Gas Ionization
2.3. Parallel Accelerator and Molecular Mass Spectrometry (PAMMS)
2.4. CRDS
3. Applications
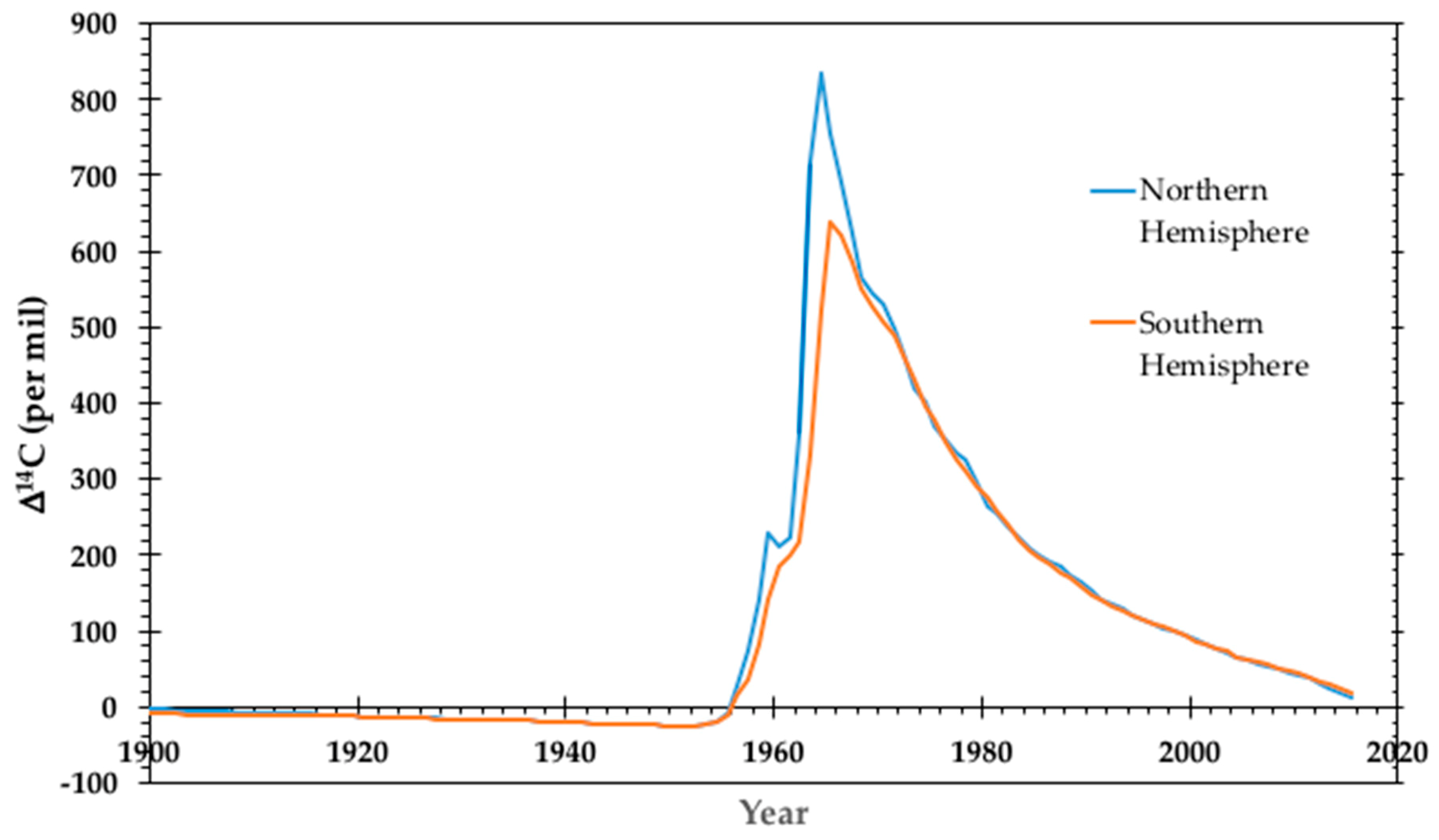
3.1. Radiocarbon Dating
3.2. Bomb Pulse Dating
3.2.1. Carbon Source Determination
3.2.2. Structural and Pathological Protein Dating
3.2.3. Cell Lifetime and Turnover
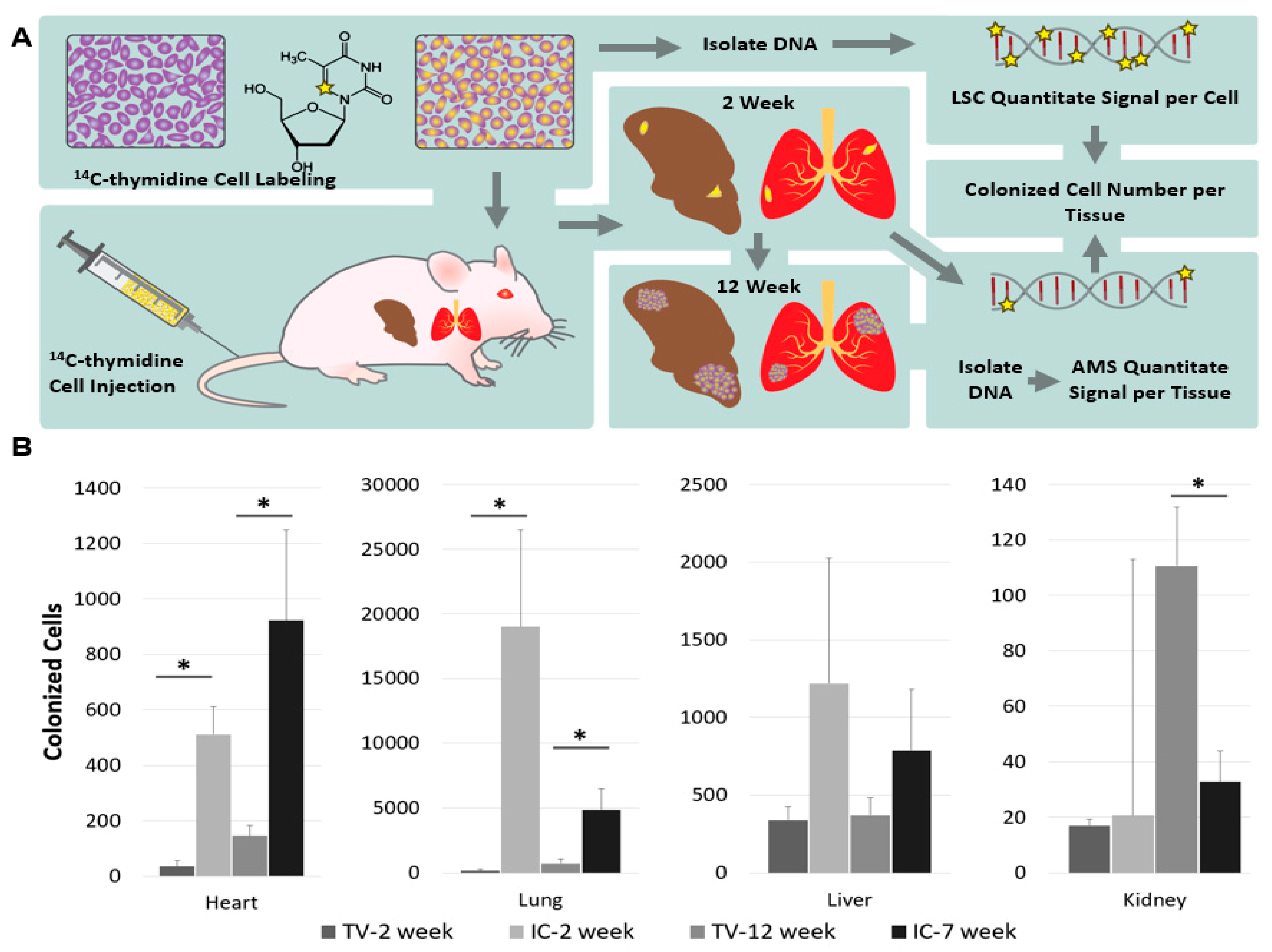
3.3. Tracking the Fate of Cells Labeled with [14C]Thymidine
3.4. Low Dose Toxicity
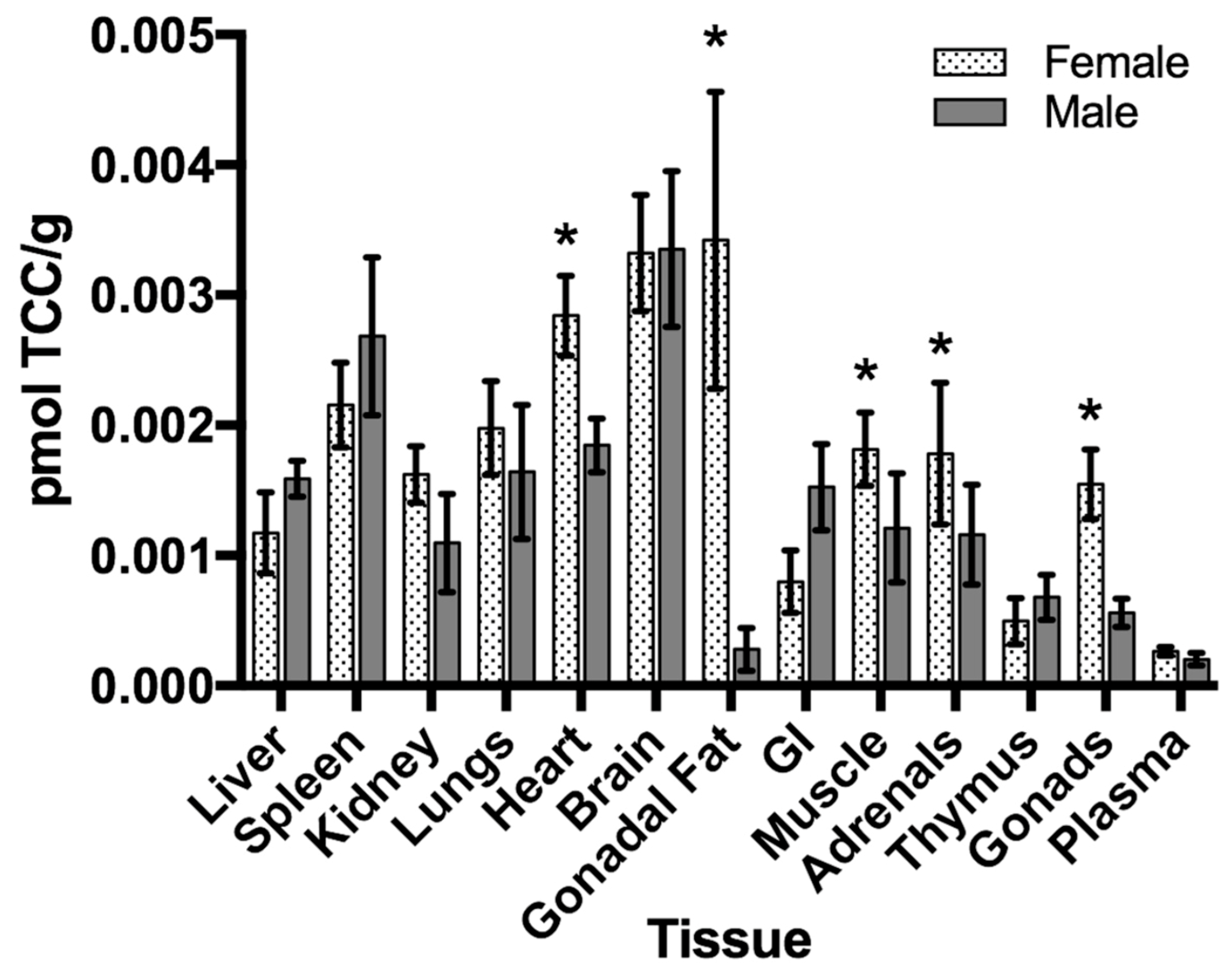
3.4.1. Naphthalene
3.4.2. Triclocarban
3.4.3. Benzo[a]pyrene
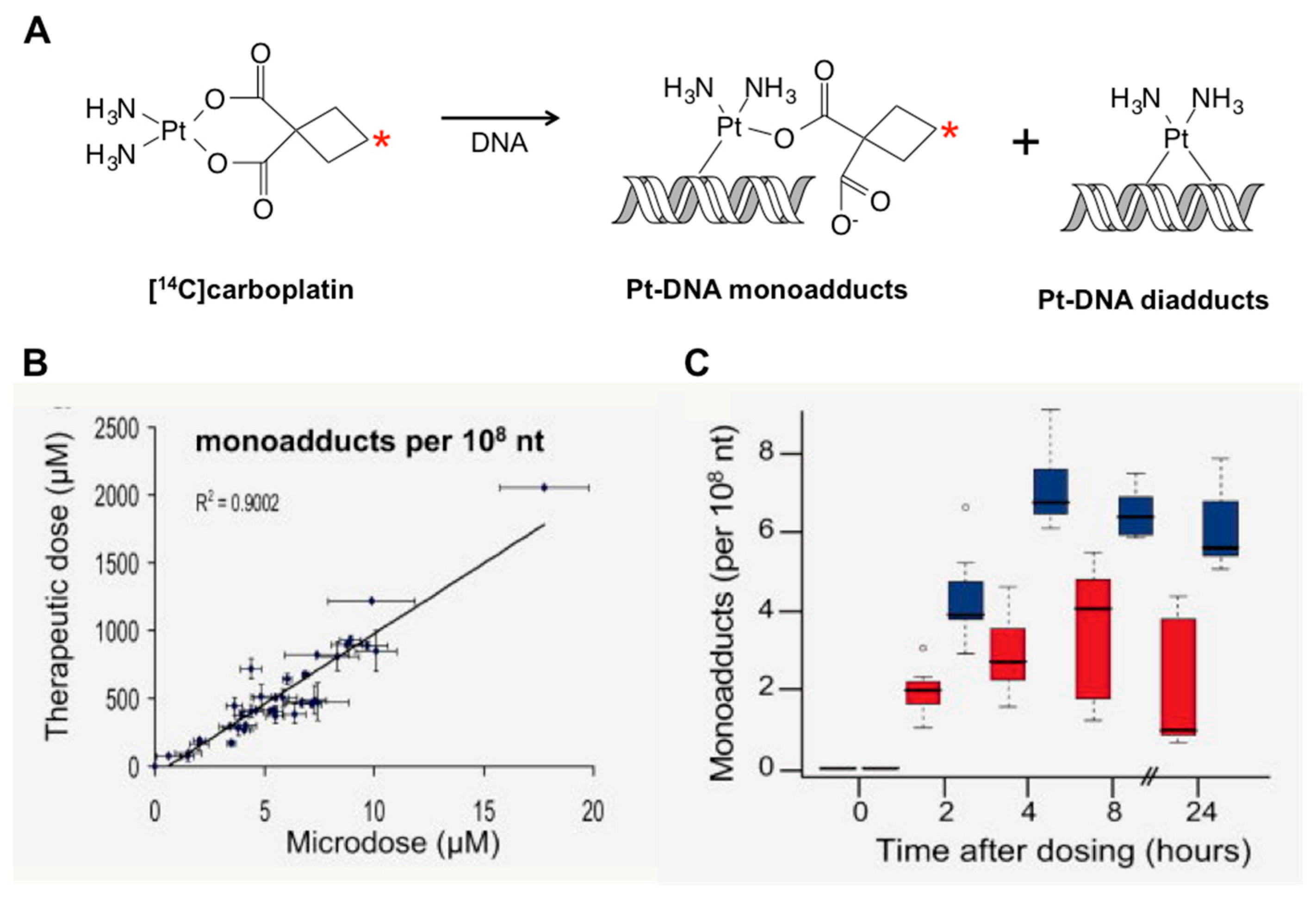
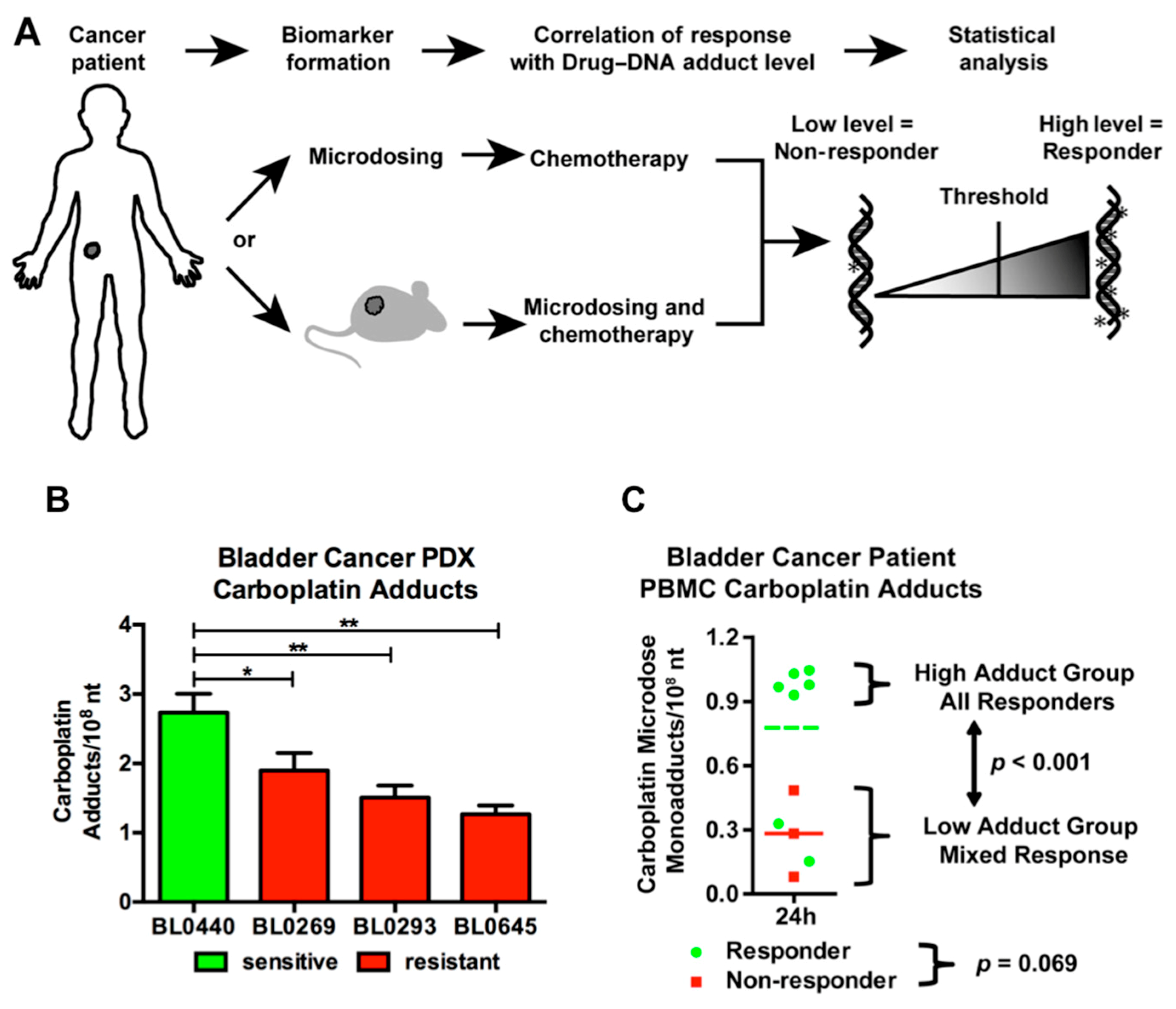
3.5. Diagnostic Microdosing: Using Drug-DNA Adducts as Biomarkers of Chemotherapy Response
3.5.1. Predicting Response to Platinum-Based Therapy with Microdosing
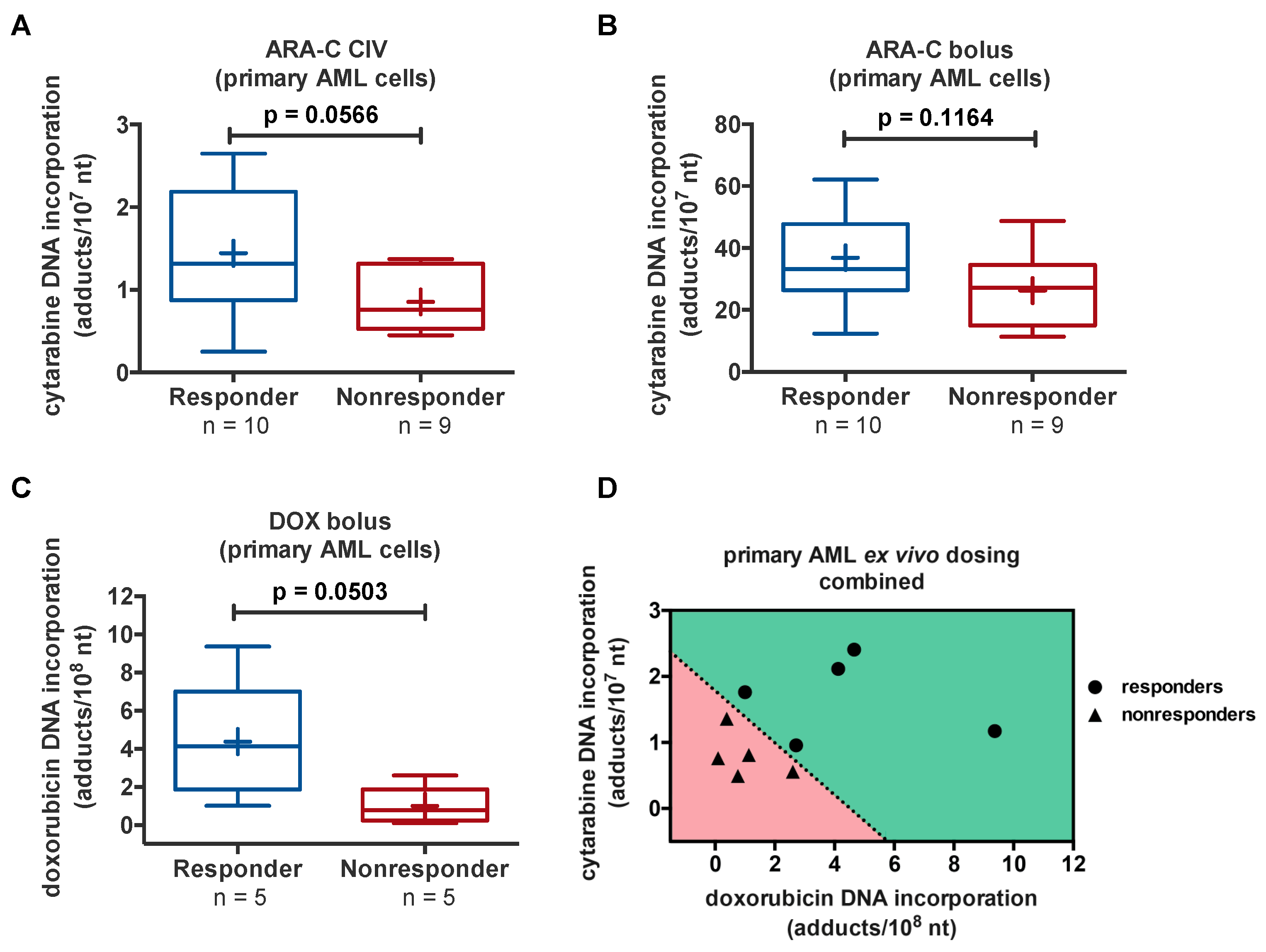
3.5.2. Ex Vivo Diagnostic Microdosing for Predicting Response to 7 + 3 in AML Patients
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Abramson, F.P. Crims—Chemical-Reaction Interface Mass-Spectrometry. Mass Spectrom. Rev. 1994, 13, 341–356. [Google Scholar] [CrossRef]
- Rosler, H. The Impact of W. K. Rontgen’s Discovery on the Use of Internalizable Sources of Ionizing Energy in Diagnostic and Therapeutic Nuclear-Medicine. Experientia 1995, 51, 686–702. [Google Scholar] [CrossRef]
- Devries, R.A.; Debruin, M.; Marx, J.J.M.; Vandewiel, A. Radioisotopic Labels for Blood-Cell Survival Studies—A Review. Nucl. Med. Biol. 1993, 20, 809–817. [Google Scholar] [CrossRef]
- Young, V.R.; Ajami, A. Isotopes in nutrition research. Proc. Nutr. Soc. 1999, 58, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.; Neuenhofer, S. Luminescent Labels—More Than Just an Alternative to Radioisotopes. Angew. Chem. Int. Ed. 1994, 33, 1044–1072. [Google Scholar] [CrossRef]
- Garner, R.C.; Barker, J.; Flavell, C.; Garner, J.V.; Whattam, M.; Young, G.C.; Cussans, N.; Jezequel, S.; Leong, D. A validation study comparing accelerator MS and liquid scintillation counting for analysis of 14C-labelled drugs in plasma, urine and faecal extracts. J. Pharm. Biomed. Anal. 2000, 24, 197–209. [Google Scholar] [CrossRef]
- Turteltaub, K.W.; Vogel, J.S. Bioanalytical applications of accelerator mass spectrometry for pharmaceutical research. Curr. Pharm. Des. 2000, 6, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.S.; Turteltaub, K.W.; Finkel, R.; Nelson, D.E. Accelerator mass spectrometry. Anal. Chem. 1995, 67, 353A–359A. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.S.; Southon, J.R.; Nelson, D.E.; Brown, T.A. Performance of Catalytically Condensed Carbon for Use in Accelerator Mass-Spectrometry. Nucl. Instrum. Methods B 1984, 5, 289–293. [Google Scholar] [CrossRef]
- Vogel, J.S.; Nelson, D.E.; Southon, J.R. C-14 Background Levels in an Accelerator Mass-Spectrometry System. Radiocarbon 1987, 29, 323–333. [Google Scholar] [CrossRef]
- Vogel, J.S.; Southon, J.R.; Nelson, D.E. Catalyst and Binder Effects in the Use of Filamentous Graphite for Ams. Nucl. Instrum. Methods B 1987, 29, 50–56. [Google Scholar] [CrossRef]
- Vogel, J.S. Rapid Production of Graphite without Contamination for Biomedical Ams. Radiocarbon 1992, 34, 344–350. [Google Scholar] [CrossRef]
- Wilson, A.T. A Simple Technique for Converting CO2 to AMS Target Graphite. Radiocarbon 1992, 34, 318–320. [Google Scholar] [CrossRef]
- Ognibene, T.J.; Bench, G.; Vogel, J.S.; Peaslee, G.F.; Murov, S. A high-throughput method for the conversion of CO2 obtained from biochemical samples to graphite in septa-sealed vials for quantification of 14C via accelerator mass spectrometry. Anal. Chem. 2003, 75, 2192–2196. [Google Scholar] [CrossRef] [PubMed]
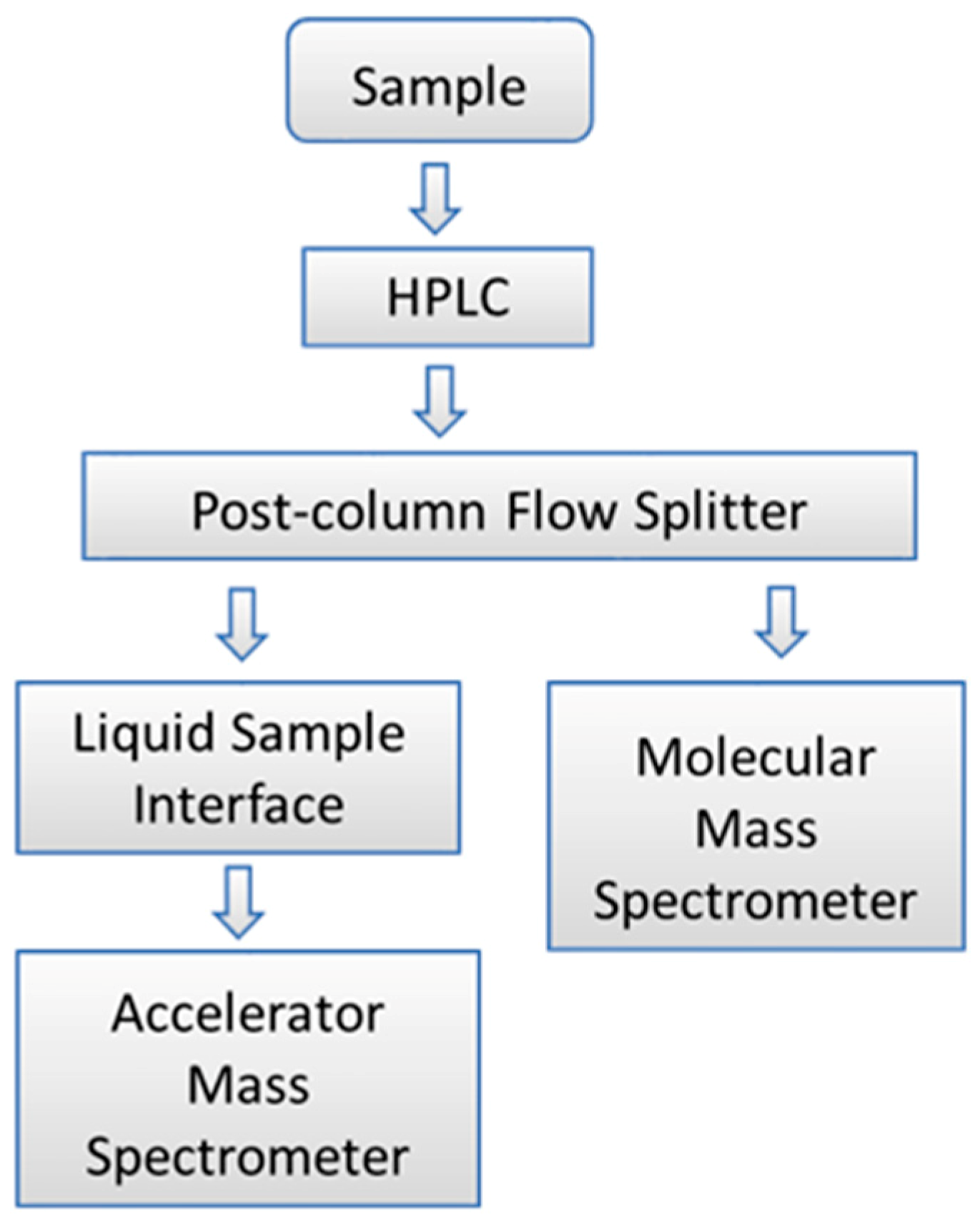
- Thomas, A.T.; Ognibene, T.; Daley, P.; Turteltaub, K.; Radousky, H.; Bench, G. Ultrahigh efficiency moving wire combustion interface for online coupling of high-performance liquid chromatography (HPLC). Anal. Chem. 2011, 83, 9413–9417. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.T.; Stewart, B.J.; Ognibene, T.J.; Turteltaub, K.W.; Bench, G. Directly coupled high-performance liquid chromatography-accelerator mass spectrometry measurement of chemically modified protein and peptides. Anal. Chem. 2013, 85, 3644–3650. [Google Scholar] [CrossRef]
- Ognibene, T.J.; Thomas, A.T.; Daley, P.F.; Bench, G.; Turteltaub, K.W. An Interface for the Direct Coupling of Small Liquid Samples to AMS. Nucl. Instrum. Methods Phys. Res. B 2015, 361, 173–177. [Google Scholar] [CrossRef]
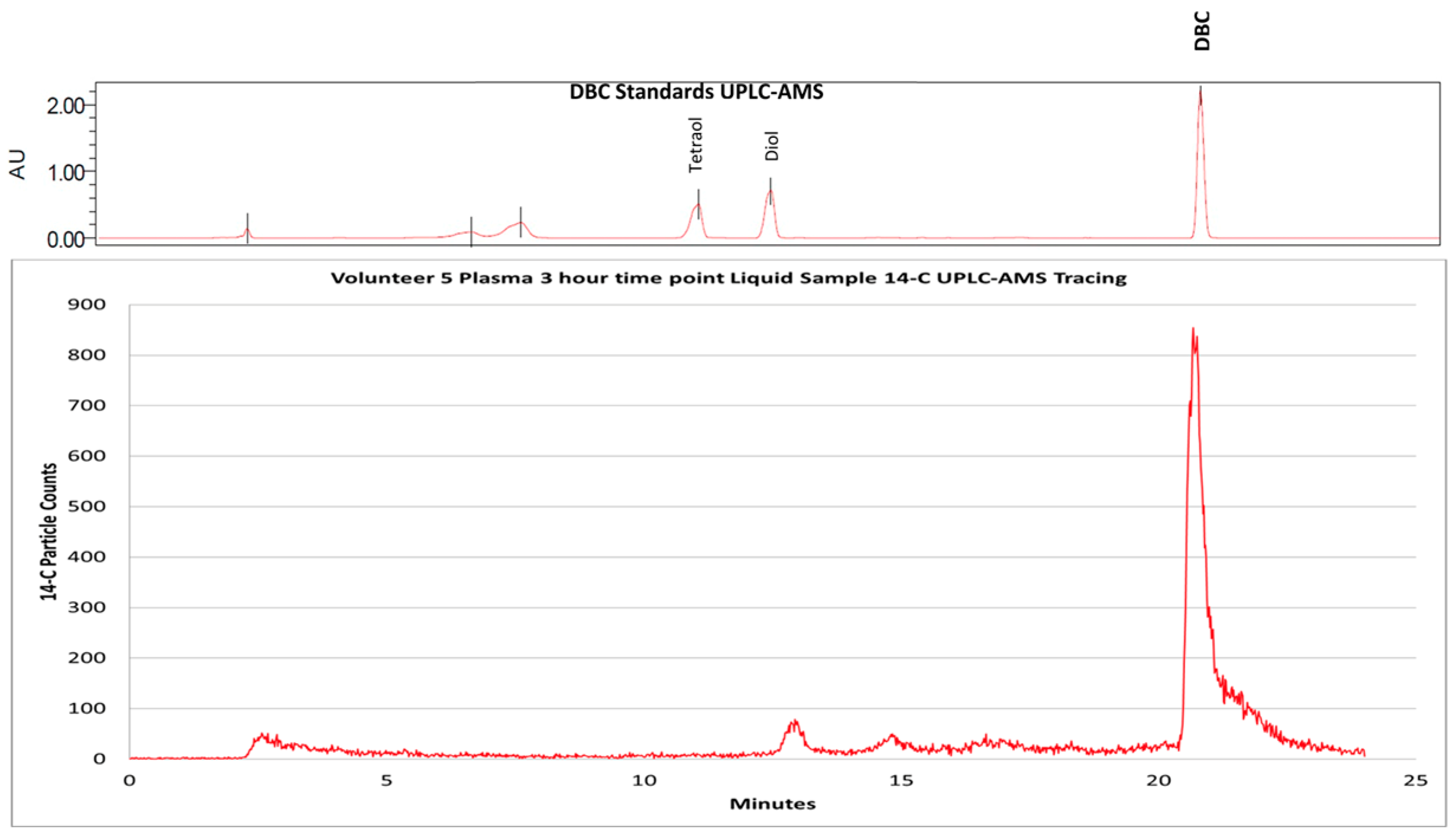
- Madeen, E.P.; Ognibene, T.J.; Corley, R.A.; McQuistan, T.J.; Henderson, M.C.; Baird, W.M.; Bench, G.; Turteltaub, K.W.; Williams, D.E. Human Microdosing with Carcinogenic Polycyclic Aromatic Hydrocarbons: In Vivo Pharmacokinetics of Dibenzo[def,p]chrysene and Metabolites by UPLC Accelerator Mass Spectrometry. Chem. Res. Toxicol. 2016, 29, 1641–1650. [Google Scholar] [CrossRef]
- Madeen, E.; Siddens, L.K.; Uesugi, S.; McQuistan, T.; Corley, R.A.; Smith, J.; Waters, K.M.; Tilton, S.C.; Anderson, K.A.; Ognibene, T.; et al. Toxicokinetics of benzo [a] pyrene in humans: Extensive metabolism as determined by UPLC-accelerator mass spectrometry following oral micro-dosing. Toxicol. Appl. Pharm. 2019, 364, 97–105. [Google Scholar] [CrossRef]
- van Duijn, E.; Sandman, H.; Grossouw, D.; Mocking, J.A.J.; Coulier, L.; Vaes, W.H.J. Automated Combustion Accelerator Mass Spectrometry for the Analysis of Biomedical Samples in the Low Attomole Range. Anal. Chem. 2014, 86, 7635–7641. [Google Scholar] [CrossRef]
- Ognibene, T.J.; Salazar, G.A. Installation of hybrid ion source on the 1-MV LLNL BioAMS spectrometer. Nucl. Instrum. Methods B 2013, 294, 311–314. [Google Scholar] [CrossRef]
- Sacks, G.L.; Derry, L.A.; Brenna, J.T. Elemental speciation by parallel elemental and molecular mass spectrometry and peak profile matching. Anal. Chem. 2006, 78, 8445–8455. [Google Scholar] [CrossRef] [PubMed]
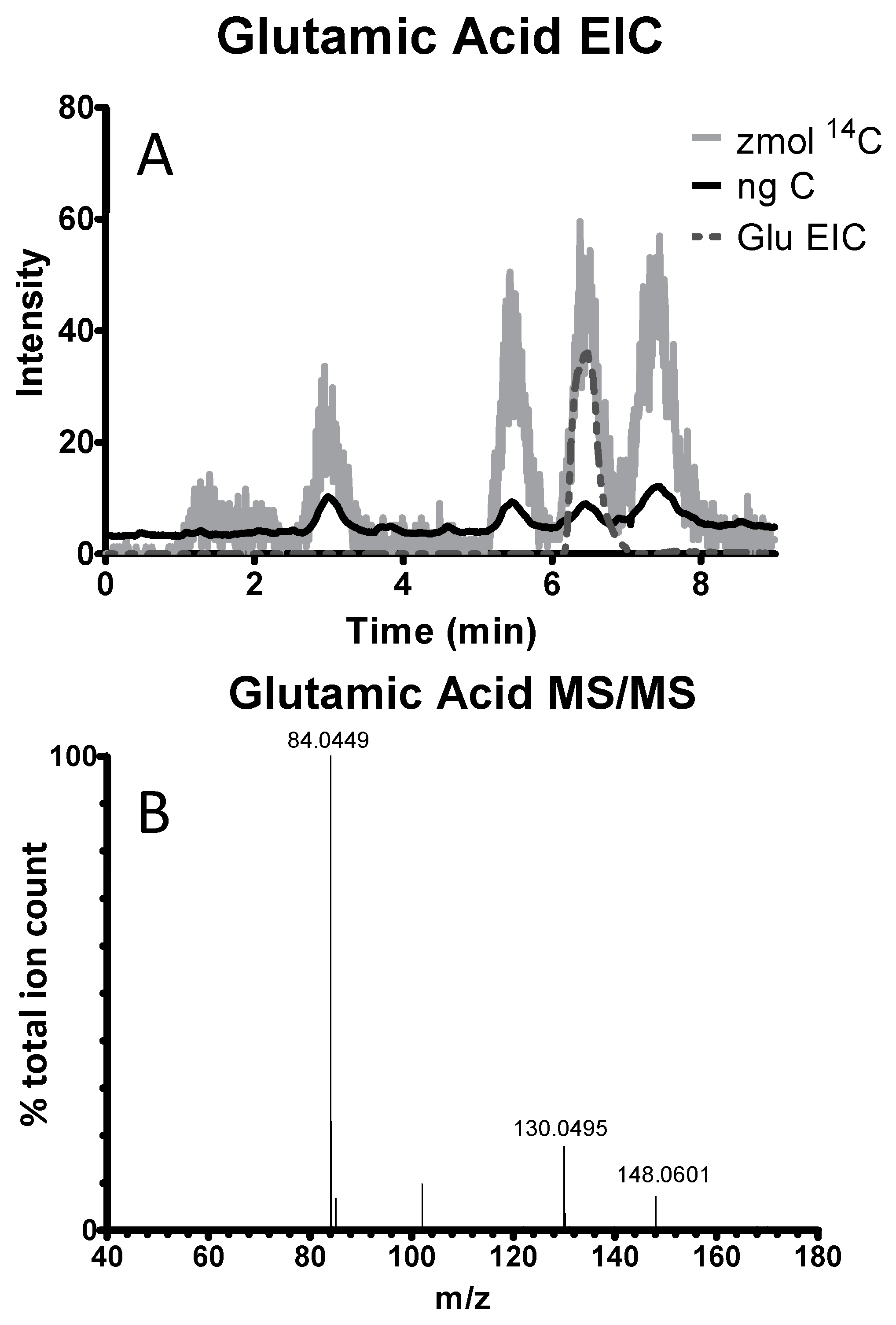
- Vogel, J.S.; Palmblad, N.M.; Ognibene, T.; Kabir, M.M.; Buchholz, B.A.; Bench, G. Biochemical paths in humans and cells: Frontiers of AMS bioanalysis. Nucl. Instrum. Methods B 2007, 259, 745–751. [Google Scholar] [CrossRef]
- Labrie, D.; Reid, J. Radiocarbon Dating by Infrared-Laser Spectroscopy—A Feasibility Study. Appl. Phys. 1981, 24, 381–386. [Google Scholar] [CrossRef]
- Murnick, D.E.; Dogru, O.; Ilkmen, E. Intracavity optogalvanic spectroscopy. An analytical technique for 14C analysis with subattomole sensitivity. Anal. Chem. 2008, 80, 4820–4824. [Google Scholar] [CrossRef]
- Galli, I.; Pastor, P.C.; Di Lonardo, G.; Fusina, L.; Giusfredi, G.; Mazzotti, D.; Tamassia, F.; De Natale, P. The ν3 band of 14C16O2 molecule measured by optical-frequency-comb-assisted cavity ring-down spectroscopy. Mol. Phys. 2011, 109, 2267–2272. [Google Scholar] [CrossRef]
- Genoud, G.; Vainio, M.; Phillips, H.; Dean, J.; Merimaa, M. Radiocarbon dioxide detection based on cavity ring-down spectroscopy and a quantum cascade laser. Opt. Lett. 2015, 40, 1342–1345. [Google Scholar] [CrossRef] [PubMed]
- McCartt, A.D.; Ognibene, T.; Bench, G.; Turteltaub, K. Measurements of Carbon-14 with Cavity Ring-Down Spectroscopy. Nucl. Instrum. Methods Phys. Res. B 2015, 361, 277–280. [Google Scholar] [CrossRef]
- Galli, I.; Bartalini, S.; Ballerini, R.; Barucci, M.; Cancio, P.; De Pas, M.; Giusfredi, G.; Mazzotti, D.; Akikusa, N.; De Natale, P. Spectroscopic detection of radiocarbon dioxide at parts-per-quadrillion sensitivity. Optica 2016, 3, 385–388. [Google Scholar] [CrossRef]
- McCartt, A.D.; Ognibene, T.J.; Bench, G.; Turteltaub, K.W. Quantifying Carbon-14 for Biology Using Cavity Ring-Down Spectroscopy. Anal. Chem. 2016, 88, 8714–8719. [Google Scholar] [CrossRef]
- Fleisher, A.J.; Long, D.A.; Liu, Q.N.; Gameson, L.; Hodges, J.T. Optical Measurement of Radiocarbon below Unity Fraction Modern by Linear Absorption Spectroscopy. J. Phys. Chem. Lett. 2017, 8, 4550–4556. [Google Scholar] [CrossRef]
- Kratochwil, N.A.; Dueker, S.R.; Muri, D.; Senn, C.; Yoon, H.; Yu, B.Y.; Lee, G.H.; Dong, F.; Otteneder, M.B. Nanotracing and cavity-ring down spectroscopy: A new ultrasensitive approach in large molecule drug disposition studies. PLoS ONE 2018, 13, e0205435. [Google Scholar] [CrossRef]
- Taylor, R.E. Dating Techniques in Archaeology and Paleoanthropology. Anal. Chem. 1987, 59, A317. [Google Scholar] [CrossRef]
- Graven, H.; Allison, C.E.; Etheridge, D.M.; Hammer, S.; Keeling, R.F.; Levin, I.; Meijer, H.A.J.; Rubino, M.; Tans, P.P.; Trudinger, C.M.; et al. Compiled records of carbon isotopes in atmospheric CO2 for historical simulations in CMIP6. Geosci. Model Dev. 2017, 10, 4405–4417. [Google Scholar] [CrossRef]
- Buchholz, B.A.; Sarachine, M.J.; Zermeno, P. Establishing Natural Product Content with Natural Radiocarbon Signature. In Progress in Authentication of Food and Wine; Ebeler, S.E., Takeoka, G.R., Winterhalter, P., Eds.; ACS Books: Washington, DC, USA, 2011; p. 27. [Google Scholar]
- Nelson, M.A.; Ondov, J.M.; VanDerveer, M.C.; Buchholz, B.A. Contemporary Fraction of Bis(2-Ethylhexyl) Phthalate in Stilton Cheese by Accelerator Mass Spectrometry. Radiocarbon 2013, 55, 686–697. [Google Scholar] [CrossRef]
- Tong, T.; Ondov, J.M.; Buchholz, B.A.; VanDerveer, M.C. Contemporary carbon content of bis (2-ethylhexyl) phthalate in butter. Food Chem. 2016, 190, 1064–1068. [Google Scholar] [CrossRef]
- Stuiver, M.; Polach, H.A. Reporting of C-14 Data—Discussion. Radiocarbon 1977, 19, 355–363. [Google Scholar] [CrossRef]
- Hedges, R.E.M.; Clement, J.G.; Thomas, C.D.L.; O’Connell, T.C. Collagen turnover in the adult femoral mid-shaft: Modeled from anthropogenic radiocarbon tracer measurements. Am. J. Phys. Anthropol. 2007, 133, 808–816. [Google Scholar] [CrossRef]
- Buchholz, B.A.; Alkass, K.; Druid, H.; Spalding, K.L. Bomb Pulse Radiocarbon Dating of Skeletal Tissues. New Perspect. Forensic Hum. Skelet. Identif. 2018, 185–196. [Google Scholar] [CrossRef]
- Shapiro, S.D.; Endicott, S.K.; Province, M.A.; Pierce, J.A.; Campbell, E.J. Marked Longevity of Human Lung Parenchymal Elastic Fibers Deduced from Prevalence of D-Aspartate and Nuclear-Weapons Related Radiocarbon. J. Clin. Invest. 1991, 87, 1828–1834. [Google Scholar] [CrossRef] [PubMed]
- Heinemeier, K.M.; Schjerling, P.; Heinemeier, J.; Magnusson, S.P.; Kjaer, M. Lack of tissue renewal in human adult Achilles tendon is revealed by nuclear bomb 14C. FASEB J. 2013, 27, 2074–2079. [Google Scholar] [CrossRef]
- Heinemeier, K.M.; Schjerling, P.; Heinemeier, J.; Moller, M.B.; Krogsgaard, M.R.; Grum-Schwensen, T.; Petersen, M.M.; Kjaer, M. Radiocarbon dating reveals minimal collagen turnover in both healthy and osteoarthritic human cartilage. Sci. Transl. Med. 2016, 8, 346ra90. [Google Scholar] [CrossRef]
- Lynnerup, N.; Kjeldsen, H.; Heegaard, S.; Jacobsen, C.; Heinemeier, J. Radiocarbon Dating of the Human Eye Lens Crystallines Reveal Proteins without Carbon Turnover throughout Life. PLoS ONE 2008, 3, e1529. [Google Scholar] [CrossRef]
- Stewart, D.N.; Lango, J.; Nambiar, K.P.; Falso, M.J.S.; FitzGerald, P.G.; Rocke, D.M.; Hammock, B.D.; Buchholz, B.A. Carbon turnover in the water-soluble protein of the adult human lens. Mol. Vis. 2013, 19, 463–475. [Google Scholar]
- Lovell, M.A.; Robertson, J.D.; Buchholz, B.A.; Xie, C.S.; Markesbery, W.R. Use of bomb pulse carbon-14 to age senile plaques and neurofibrillary tangles in Alzheimer’s disease. Neurobiol. Aging 2002, 23, 179–186. [Google Scholar] [CrossRef]
- Goncalves, I.; Stenstrom, K.; Skog, G.; Mattsson, S.; Nitulescu, M.; Nilsson, J. Dating components of human atherosclerotic plaques. Eur. Heart J. 2010, 31, 794. [Google Scholar]
- Hagg, S.; Salehpour, M.; Noori, P.; Lundstrom, J.; Possnert, G.; Takolander, R.; Konrad, P.; Rosfors, S.; Ruusalepp, A.; Skogsberg, J.; et al. Carotid Plaque Age Is a Feature of Plaque Stability Inversely Related to Levels of Plasma Insulin. PLoS ONE 2011, 6, e18248. [Google Scholar] [CrossRef]
- Etminan, N.; Dreier, R.; Buchholz, B.A.; Beseoglu, K.; Bruckner, P.; Matzenauer, C.; Torner, J.C.; Brown, R.D.; Steiger, H.J.; Haggi, D.; et al. Age of Collagen in Intracranial Saccular Aneurysms. Stroke 2014, 45, 1757–1763. [Google Scholar] [CrossRef]
- Etminan, N.; Dreier, R.; Buchholz, B.A.; Bruckner, P.; Steiger, H.J.; Hanggi, D.; Macdonald, R.L. Exploring the Age of Intracranial Aneurysms Using Carbon Birth Dating Preliminary Results. Stroke 2013, 44, 799–802. [Google Scholar] [CrossRef]
- Spalding, K.L.; Bhardwaj, R.D.; Buchholz, B.A.; Druid, H.; Frisen, J. Retrospective birth dating of cells in humans. Cell 2005, 122, 133–143. [Google Scholar] [CrossRef]
- Bhardwaj, R.D.; Curtis, M.A.; Spalding, K.L.; Buchholz, B.A.; Fink, D.; Bjork-Eriksson, T.; Nordborg, C.; Gage, F.H.; Druid, H.; Eriksson, P.S.; et al. Neocortical neurogenesis in humans is restricted to development. Proc. Natl. Acad. Sci. USA 2006, 103, 12564–12568. [Google Scholar] [CrossRef] [PubMed]
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Bostrom, E.; Westerlund, I.; Vial, C.; Buchholz, B.A.; et al. Dynamics of Hippocampal Neurogenesis in Adult Humans. Cell 2013, 153, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Liebl, J.; Bernard, S.; Alkass, K.; Yeung, M.S.; Steier, P.; Kutschera, W.; Johnson, L.; Landen, M.; Druid, H.; et al. The age of olfactory bulb neurons in humans. Neuron 2012, 74, 634–639. [Google Scholar] [CrossRef]
- Yeung, M.S.; Zdunek, S.; Bergmann, O.; Bernard, S.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Brundin, L.; et al. Dynamics of oligodendrocyte generation and myelination in the human brain. Cell 2014, 159, 766–774. [Google Scholar] [CrossRef]
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Naslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783–787. [Google Scholar] [CrossRef]
- Arner, P.; Bernard, S.; Salehpour, M.; Possnert, G.; Liebl, J.; Steier, P.; Buchholz, B.A.; Eriksson, M.; Arner, E.; Hauner, H.; et al. Dynamics of human adipose lipid turnover in health and metabolic disease. Nature 2011, 478, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabe-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Perl, S.; Kushner, J.A.; Buchholz, B.A.; Meeker, A.K.; Stein, G.M.; Hsieh, M.; Kirby, M.; Pechhold, S.; Liu, E.H.; Harlan, D.M.; et al. Significant human beta-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J. Clin. Endocrinol. Metab. 2010, 95, E234–E239. [Google Scholar] [CrossRef]
- Landsverk, O.J.; Snir, O.; Casado, R.B.; Richter, L.; Mold, J.E.; Reu, P.; Horneland, R.; Paulsen, V.; Yaqub, S.; Aandahl, E.M.; et al. Antibody-secreting plasma cells persist for decades in human intestine. J. Exp. Med. 2017, 214, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Maze, I.; Wenderski, W.; Noh, K.M.; Bagot, R.C.; Tzavaras, N.; Purushothaman, I.; Elsasser, S.J.; Guo, Y.; Ionete, C.; Hurd, Y.L.; et al. Critical Role of Histone Turnover in Neuronal Transcription and Plasticity. Neuron 2015, 87, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Hum, N.R.; Martin, K.A.; Malfatti, M.A.; Haack, K.; Buchholz, B.A.; Loots, G.G. Tracking Tumor Colonization in Xenograft Mouse Models Using Accelerator Mass Spectrometry. Sci. Rep. 2018, 8, 15013. [Google Scholar] [CrossRef]
- Orjuela, M.A.; Liu, X.; Miller, R.L.; Warburton, D.; Tang, D.; Jobanputra, V.; Hoepner, L.; Suen, I.H.; Diaz-Carreno, S.; Li, Z.; et al. Urinary naphthol metabolites and chromosomal aberrations in 5-year-old children. Cancer Epidemiol. Biomarkers Prev. 2012, 21, 1191–1202. [Google Scholar] [CrossRef]
- Abdo, K.; Eustic, S.; McDonald, M.; Jokinen, M.; Adkins, B.; Haseman, J. Naphthalene: A respiratory tract toxicant and carcinogen for mice. Inhal. Toxicol. 1992, 4, 393–409. [Google Scholar] [CrossRef]
- North, D.W.; Abdo, K.M.; Benson, J.M.; Dahl, A.R.; Morris, J.B.; Renne, R.; Witschi, H. A review of whole animal bioassays of the carcinogenic potential of naphthalene. Regul. Toxicol. Pharm. 2008, 51, S6–S14. [Google Scholar] [CrossRef]
- Buchholz, B.A.; Haack, K.W.; Sporty, J.L.; Buckpitt, A.R.; Morin, D. Free flow electrophoresis separation and AMS quantitation of 14C-naphthalene-protein adducts. Nucl. Instrum. Methods B 2010, 268, 1324–1327. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, B.A.; Carratt, S.A.; Kuhn, E.A.; Collette, N.M.; Ding, X.X.; Van Winkle, L.S. Naphthalene DNA adduct formation and tolerance in the lung. Nucl. Instrum. Methods B 2019, 438, 119–123. [Google Scholar] [CrossRef]
- Van Winkle, L.S.; Kelty, J.S.; Plopper, C.G. Preparation of Specific Compartments of the Lungs for Pathologic and Biochemical Analysis of Toxicologic Responses. Curr. Protoc. Toxicol. 2017, 71, 24.5.1–24.5.26. [Google Scholar] [CrossRef]
- Morris, J.B. Nasal dosimetry of inspired naphthalene vapor in the male and female B6C3F1 mouse. Toxicology 2013, 309, 66–72. [Google Scholar] [CrossRef]
- Enright, H.A.; Falso, M.J.S.; Malfatti, M.A.; Lao, V.; Kuhn, E.A.; Hum, N.; Shi, Y.; Sales, A.P.; Haack, K.W.; Kulp, K.S.; et al. Maternal exposure to an environmentally relevant dose of triclocarban results in perinatal exposure and potential alterations in offspring development in the mouse model. PLoS ONE 2017, 12, e0181996. [Google Scholar] [CrossRef]
- Halden, R.U. On the need and speed of regulating triclosan and triclocarban in the United States. Environ. Sci. Technol. 2014, 48, 3603–3611. [Google Scholar] [CrossRef]
- Coogan, M.A.; La Point, T.W. Snail bioaccumulation of triclocarban, triclosan, and methyltriclosan in a North Texas, USA, stream affected by wastewater treatment plant runoff. Environ. Toxicol. Chem. 2008, 27, 1788–1793. [Google Scholar] [CrossRef] [PubMed]
- Geiss, C.; Ruppert, K.; Heidelbach, T.; Oehlmann, J. The antimicrobial agents triclocarban and triclosan as potent modulators of reproduction in Potamopyrgus antipodarum (Mollusca: Hydrobiidae). J. Environ. Sci. Health A Tox. Hazard Subst. Environ. Eng. 2016, 51, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- IARC. Some Non-heterocyclic Polycyclic Aromatic Hydrocarbons and some Related Exposures. Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2010. [Google Scholar]
- World Health Organization (WHO); International Programme on Chemical Safety. Selected Non-Heterocyclic Polycyclic Aromatic Hydrocarbons; Environmental Health Criteria 202; WHO: Geneva, Switzerland, 1998. [Google Scholar]
- EPA. Toxicological Review of Benzo[a]pyrene, Integrated Risk Information System, National Center for Environmental Assessment; Integrated Risk Information System, National Center for Environmental Assessment, EPA: Washington, DC, USA, 2017.
- Abadin, H.G. The toxicological profile program at ATSDR. J. Environ. Health 2013, 75, 42–43. [Google Scholar] [PubMed]
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef] [PubMed]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [PubMed]
- Boocock, D.J.; Brown, K.; Gibbs, A.H.; Sanchez, E.; Turteltaub, K.W.; White, I.N. Identification of human CYP forms involved in the activation of tamoxifen and irreversible binding to DNA. Carcinogenesis 2002, 23, 1897–1901. [Google Scholar] [CrossRef]
- Martin, E.A.; Brown, K.; Gaskell, M.; Al-Azzawi, F.; Garner, R.C.; Boocock, D.J.; Mattock, E.; Pring, D.W.; Dingley, K.; Turteltaub, K.W.; et al. Tamoxifen DNA damage detected in human endometrium using accelerator mass spectrometry. Cancer Res. 2003, 63, 8461–8465. [Google Scholar]
- Hah, S.S.; Sumbad, R.A.; de Vere White, R.W.; Turteltaub, K.W.; Henderson, P.T. Characterization of oxaliplatin-DNA adduct formation in DNA and differentiation of cancer cell drug sensitivity at microdose concentrations. Chem. Res. Toxicol. 2007, 20, 1745–1751. [Google Scholar] [CrossRef]
- Brown, K.; Tompkins, E.M.; Boocock, D.J.; Martin, E.A.; Farmer, P.B.; Turteltaub, K.W.; Ubick, E.; Hemingway, D.; Horner-Glister, E.; White, I.N. Tamoxifen forms DNA adducts in human colon after administration of a single [14C]-labeled therapeutic dose. Cancer Res. 2007, 67, 6995–7002. [Google Scholar] [CrossRef] [PubMed]
- Hah, S.S.; Henderson, P.T.; Turteltaub, K.W. Towards biomarker-dependent individualized chemotherapy: Exploring cell-specific differences in oxaliplatin-DNA adduct distribution using accelerator mass spectrometry. Bioorganic Med. Chem. Lett. 2010, 20, 2448–2451. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, H.; Malfatti, M.; de Vere White, R.; Lara, P.N., Jr.; Turteltaub, K.; Henderson, P.; Pan, C.X. Gemcitabine causes minimal modulation of carboplatin-DNA monoadduct formation and repair in bladder cancer cells. Chem. Res. Toxicol. 2010, 23, 1653–1655. [Google Scholar] [CrossRef] [PubMed]
- Henderson, P.T.; Li, T.; He, M.; Zhang, H.; Malfatti, M.; Gandara, D.; Grimminger, P.P.; Danenberg, K.D.; Beckett, L.; de Vere White, R.W.; et al. A microdosing approach for characterizing formation and repair of carboplatin-DNA monoadducts and chemoresistance. Int. J. Cancer 2011, 129, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Pan, A.W.; Lin, T.Y.; Zhang, H.; Malfatti, M.; Turteltaub, K.; Henderson, P.T.; Pan, C.X. Paclitaxel Enhances Carboplatin-DNA Adduct Formation and Cytotoxicity. Chem. Res. Toxicol. 2015, 28, 2250–2252. [Google Scholar] [CrossRef] [PubMed]
- Scharadin, T.M.; Zhang, H.; Zimmermann, M.; Wang, S.; Malfatti, M.A.; Cimino, G.D.; Turteltaub, K.; de Vere White, R.; Pan, C.X.; Henderson, P.T. Diagnostic Microdosing Approach to Study Gemcitabine Resistance. Chem. Res. Toxicol. 2016, 29, 1843–1848. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, H.; Scharadin, T.M.; Zimmermann, M.; Hu, B.; Pan, A.W.; Vinall, R.; Lin, T.Y.; Cimino, G.; Chain, P.; et al. Molecular Dissection of Induced Platinum Resistance through Functional and Gene Expression Analysis in a Cell Culture Model of Bladder Cancer. PLoS ONE 2016, 11, e0146256. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Wang, S.S.; Zhang, H.; Lin, T.Y.; Malfatti, M.; Haack, K.; Ognibene, T.; Yang, H.; Airhart, S.; Turteltaub, K.W.; et al. Microdose-Induced Drug-DNA Adducts as Biomarkers of Chemotherapy Resistance in Humans and Mice. Mol. Cancer Ther. 2017, 16, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Zimmermann, M.; Zhang, H.; Lin, T.Y.; Malfatti, M.; Haack, K.; Turteltaub, K.W.; Cimino, G.D.; de Vere White, R.; Pan, C.X.; et al. A diagnostic microdosing approach to investigate platinum sensitivity in non-small cell lung cancer. Int. J. Cancer 2017, 141, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, H.; Ma, A.H.; Yu, W.; Zimmermann, M.; Yang, J.; Hwang, S.H.; Zhu, D.; Lin, T.Y.; Malfatti, M.; et al. COX-2/sEH Dual Inhibitor PTUPB Potentiates the Anti-tumor Efficacy of Cisplatin. Mol. Cancer Ther. 2017. [Google Scholar] [CrossRef]
- Scharadin, T.M.; Malfatti, M.A.; Haack, K.; Turteltaub, K.W.; Pan, C.X.; Henderson, P.T.; Jonas, B.A. Towards predicting AML patient response to 7+3 induction chemotherapy via diagnostic microdosing. Chem. Res. Toxicol. 2018. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Rai, K.R.; Holland, J.F.; Glidewell, O.J.; Weinberg, V.; Brunner, K.; Obrecht, J.P.; Preisler, H.D.; Nawabi, I.W.; Prager, D.; Carey, R.W.; et al. Treatment of acute myelocytic leukemia: A study by cancer and leukemia group B. Blood 1981, 58, 1203–1212. [Google Scholar]
- Lowenberg, B.; Downing, J.R.; Burnett, A. Acute myeloid leukemia. N. Engl. J. Med. 1999, 341, 1051–1062. [Google Scholar] [CrossRef]
- Sekeres, M.A.; Elson, P.; Kalaycio, M.E.; Advani, A.S.; Copelan, E.A.; Faderl, S.; Kantarjian, H.M.; Estey, E. Time from diagnosis to treatment initiation predicts survival in younger, but not older, acute myeloid leukemia patients. Blood 2009, 113, 28–36. [Google Scholar] [CrossRef]
- Juliusson, G.; Antunovic, P.; Derolf, A.; Lehmann, S.; Mollgard, L.; Stockelberg, D.; Tidefelt, U.; Wahlin, A.; Hoglund, M. Age and acute myeloid leukemia: Real world data on decision to treat and outcomes from the Swedish Acute Leukemia Registry. Blood 2009, 113, 4179–4187. [Google Scholar] [CrossRef]
- Kern, W.; Estey, E.H. High-dose cytosine arabinoside in the treatment of acute myeloid leukemia: Review of three randomized trials. Cancer 2006, 107, 116–124. [Google Scholar] [CrossRef]
- Willemze, R.; Suciu, S.; Meloni, G.; Labar, B.; Marie, J.P.; Halkes, C.J.; Muus, P.; Mistrik, M.; Amadori, S.; Specchia, G.; et al. High-dose cytarabine in induction treatment improves the outcome of adult patients younger than age 46 years with acute myeloid leukemia: Results of the EORTC-GIMEMA AML-12 trial. J. Clin. Oncol. 2014, 32, 219–228. [Google Scholar] [CrossRef]
- Weick, J.K.; Kopecky, K.J.; Appelbaum, F.R.; Head, D.R.; Kingsbury, L.L.; Balcerzak, S.P.; Bickers, J.N.; Hynes, H.E.; Welborn, J.L.; Simon, S.R.; et al. A randomized investigation of high-dose versus standard-dose cytosine arabinoside with daunorubicin in patients with previously untreated acute myeloid leukemia: A Southwest Oncology Group study. Blood 1996, 88, 2841–2851. [Google Scholar]
- Bishop, J.F.; Matthews, J.P.; Young, G.A.; Szer, J.; Gillett, A.; Joshua, D.; Bradstock, K.; Enno, A.; Wolf, M.M.; Fox, R.; et al. A randomized study of high-dose cytarabine in induction in acute myeloid leukemia. Blood 1996, 87, 1710–1717. [Google Scholar]
- Cutts, S.M.; Swift, L.P.; Pillay, V.; Forrest, R.A.; Nudelman, A.; Rephaeli, A.; Phillips, D.R. Activation of clinically used anthracyclines by the formaldehyde-releasing prodrug pivaloyloxymethyl butyrate. Mol. Cancer Ther. 2007, 6, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Major, P.P.; Egan, E.M.; Beardsley, G.P.; Minden, M.D.; Kufe, D.W. Lethality of human myeloblasts correlates with the incorporation of arabinofuranosylcytosine into DNA. Proc. Natl. Acad. Sci. USA 1981, 78, 3235–3239. [Google Scholar] [CrossRef] [PubMed]
- Kufe, D.W.; Munroe, D.; Herrick, D.; Egan, E.; Spriggs, D. Effects of 1-beta-D-arabinofuranosylcytosine incorporation on eukaryotic DNA template function. Mol. Pharmacol. 1984, 26, 128–134. [Google Scholar]
- Raza, A.; Gezer, S.; Anderson, J.; Lykins, J.; Bennett, J.; Browman, G.; Goldberg, J.; Larson, R.; Vogler, R.; Preisler, H.D. Relationship of [3H]Ara-C incorporation and response to therapy with high-dose Ara-C in AML patients: A Leukemia Intergroup study. Exp. Hematol. 1992, 20, 1194–1200. [Google Scholar]
- Gervasoni, J.E., Jr.; Fields, S.Z.; Krishna, S.; Baker, M.A.; Rosado, M.; Thuraisamy, K.; Hindenburg, A.A.; Taub, R.N. Subcellular distribution of daunorubicin in P-glycoprotein-positive and -negative drug-resistant cell lines using laser-assisted confocal microscopy. Cancer Res. 1991, 51, 4955–4963. [Google Scholar]
- Coley, H.M.; Amos, W.B.; Twentyman, P.R.; Workman, P. Examination by laser scanning confocal fluorescence imaging microscopy of the subcellular localisation of anthracyclines in parent and multidrug resistant cell lines. Br. J. Cancer 1993, 67, 1316–1323. [Google Scholar] [CrossRef]
- Swift, L.P.; Rephaeli, A.; Nudelman, A.; Phillips, D.R.; Cutts, S.M. Doxorubicin-DNA adducts induce a non-topoisomerase II-mediated form of cell death. Cancer Res. 2006, 66, 4863–4871. [Google Scholar] [CrossRef]
- Coldwell, K.E.; Cutts, S.M.; Ognibene, T.J.; Henderson, P.T.; Phillips, D.R. Detection of Adriamycin-DNA adducts by accelerator mass spectrometry at clinically relevant Adriamycin concentrations. Nucleic Acids Res. 2008, 36, e100. [Google Scholar] [CrossRef] [PubMed]
- Stornetta, A.; Zimmermann, M.; Cimino, G.D.; Henderson, P.T.; Sturla, S.J. DNA Adducts from Anticancer Drugs as Candidate Predictive Markers for Precision Medicine. Chem. Res. Toxicol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Swart, P.; Lozac’h, F.; Simon, M.; van Duijn, E.; Vaes, W.H. The impact of early human data on clinical development: There is time to win. Drug Discov. Today 2016, 21, 873–879. [Google Scholar] [CrossRef]
- Morcos, P.N.; Yu, L.; Bogman, K.; Sato, M.; Katsuki, H.; Kawashima, K.; Moore, D.J.; Whayman, M.; Nieforth, K.; Heinig, K.; et al. Absorption, distribution, metabolism and excretion (ADME) of the ALK inhibitor alectinib: Results from an absolute bioavailability and mass balance study in healthy subjects. Xenobiotica 2017, 47, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Husser, C.; Pahler, A.; Seymour, M.; Kuhlmann, O.; Schadt, S.; Zell, M. Profiling of dalcetrapib metabolites in human plasma by accelerator mass spectrometry and investigation of the free phenothiol by derivatisation with methylacrylate. J. Pharm. Biomed. Anal. 2018, 152, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Schadt, S.; Bister, B.; Chowdhury, S.K.; Funk, C.; Hop, C.; Humphreys, W.G.; Igarashi, F.; James, A.D.; Kagan, M.; Khojasteh, S.C.; et al. A Decade in the MIST: Learnings from Investigations of Drug Metabolites in Drug Development under the “Metabolites in Safety Testing” Regulatory Guidance. Drug Metab. Dispos. 2018, 46, 865–878. [Google Scholar] [CrossRef] [PubMed]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malfatti, M.A.; Buchholz, B.A.; Enright, H.A.; Stewart, B.J.; Ognibene, T.J.; McCartt, A.D.; Loots, G.G.; Zimmermann, M.; Scharadin, T.M.; Cimino, G.D.; et al. Radiocarbon Tracers in Toxicology and Medicine: Recent Advances in Technology and Science. Toxics 2019, 7, 27. https://doi.org/10.3390/toxics7020027
Malfatti MA, Buchholz BA, Enright HA, Stewart BJ, Ognibene TJ, McCartt AD, Loots GG, Zimmermann M, Scharadin TM, Cimino GD, et al. Radiocarbon Tracers in Toxicology and Medicine: Recent Advances in Technology and Science. Toxics. 2019; 7(2):27. https://doi.org/10.3390/toxics7020027
Chicago/Turabian StyleMalfatti, Michael A., Bruce A. Buchholz, Heather A. Enright, Benjamin J. Stewart, Ted J. Ognibene, A. Daniel McCartt, Gabriela G. Loots, Maike Zimmermann, Tiffany M. Scharadin, George D. Cimino, and et al. 2019. "Radiocarbon Tracers in Toxicology and Medicine: Recent Advances in Technology and Science" Toxics 7, no. 2: 27. https://doi.org/10.3390/toxics7020027
APA StyleMalfatti, M. A., Buchholz, B. A., Enright, H. A., Stewart, B. J., Ognibene, T. J., McCartt, A. D., Loots, G. G., Zimmermann, M., Scharadin, T. M., Cimino, G. D., Jonas, B. A., Pan, C.-X., Bench, G., Henderson, P. T., & Turteltaub, K. W. (2019). Radiocarbon Tracers in Toxicology and Medicine: Recent Advances in Technology and Science. Toxics, 7(2), 27. https://doi.org/10.3390/toxics7020027