1. Introduction
The catalytic carboxylation of α-oxides is one of the most effective methods for producing cyclic carbonates, which are widely used as electrolytes for Li-batteries, solvents, monomers, etc. [
1,
2]. Amongst the numerous catalysts used for carrying out these processes, from a practical point of view, zinc compounds in combination with quaternary ammonium salts ZnX
2/R
4N
+X
− (X = Hal, R = Alk) are very promising [
3,
4]. It is known that each of the components of these systems, as a rule, is a low-active catalyst in this reaction, but their combined use significantly increases the rate and ensures mild processing conditions. It should be noted that these reactions are typically studied by determining the yield of the final product, whereas the observation of changes in processing parameters (pressure and temperature) over time are usually not carried out.
In this work, the preparation of propylene carbonate (PC) by the carboxylation of propylene oxide (PO) was carried out together with the continuous automatic recording of the pressure and temperature using electronic sensors. Since the studied reaction was used to obtain the PC labeled with a stable carbon isotope
13C (an intermediate product in the synthesis of the diagnostic drug
13C-urea [
5]) [
6,
7,
8] as shown in Equation (1), the process was carried out under conditions of CO
2 deficiency (relative to PO) for the economy of the labeled raw materials. This technique allowed us to reveal several unusual features of the reaction, the study of which was the purpose of this work.
2. Materials and Methods
2.1. Chemicals
Propylene oxide, zinc bromide, tetrabutylammonium bromide, tetrabutylammonium iodide, and butyltriphenylphosphonium bromide (all 99% purity) were obtained from Acros Organics. Zinc chloride, aluminum bromide (anhydrous), aluminum chloride hexahydrate, potassium iodide, catechol, tetraethylammonium bromide, and triethanolamine were commercial chemicals (reagent grade) and were used without purification. Carbon dioxide 99.99% was obtained from NII KM (Moscow, Russia).
2.2. Catalytic Experiments
The reaction was carried out in a 200 mL stainless steel thermostatted autoclave equipped with sensors for automatic recording of temperature and pressure in 1-min increments. Automatic recording of pressure was carried out using a PD100-DI6.0-111-0.5 sensor, and the temperature was measured with a chromel-copel thermocouple. The signals of the pressure sensor and thermocouple were transmitted to a TRM 200 pressure meter and then through an AC4 automatic converter (all equipment was purchased from “Oven”, Moscow, Russia)—and finally—to the computer. The components of the catalytic system were placed into the autoclave, and after sealing, it was evacuated and propylene oxide was loaded using a siphon from a measuring vessel, and CO
2 was supplied at the required pressure, followed by the closure of the gas supply valve. Thereafter, the stirring and heating were turned on, and changes in temperature and pressure during the reaction were registered. Upon process completion (pressure drop stopping), the autoclave was cooled, the unreacted propylene oxide was removed using a water jet vacuum-pump, and the resulting crude product was unloaded. Propylene carbonate was distilled off on a rotary evaporator under vacuum at 82–84 °C/3 mm Hg (lit. m.p. 241.7 °C/760 mm Hg [
9]). The finished product had a n
D = 1.4209 (lit. n
D20 = 1.4209 [
9]) and a purity of more than 99% on the
1H NMR data in DMSO-d
6 (δ, ppm): 1.37 d (3H, CH
3,
3JCH–CH3 6.1 Hz), 4.06 m (1H, not equiv. CH
2,
2JCH2 ≈
3JCH–CH2 7.3–8.6 Hz), 4.57 t (1H, not equiv. CH
2,
2JCH2 ≈
3JCH–CH2 7.9–8.6 Hz), 4.89 m (1H, CH). To determine the yield of the propylene carbonate, based on the CO
2 loaded, the gas was loaded into the autoclave from a small cylinder and weighed with an accuracy of 0.01 g before and after the process.
In a standard experiment, ZnBr2 (33.4 mg, 0.148 mmol) and Et4NBr (122.5 mg, 0.583 mmol) were loaded into the autoclave, and after sealing and evacuating the autoclave, propylene oxide (20 mL, 16.6 g, 285.8 mmol) was added using the siphon. Then, CO2 was supplied from the weighted balloon at a pressure of 15.6 bar such that after the second weighing of the balloon, the CO2 loading was determined, which amounted to 11.92 g (6068.4 mL, 270.9 mmol). Stirring and heating to a temperature of 103 °C commenced, and during the process, changes in pressure and temperature in the autoclave were observed. After 150 min the pressure drop ceased, indicating that the reaction was complete. Next, the autoclave was cooled, and the product was isolated according to the above procedure for analysis. The rest of the experiments were carried out using the same method, the loads and the conditions of which are presented below.
2.3. Product Analysis
Analysis of the impurities content in the obtained propylene carbonate was carried out using a Crystallux-4000M gas chromatograph (from “Meta-chrom” company, Yoshkar-Ola, Russia) with a flame ionization detector and a capillary column (25 m × 0.2 mm) with a stationary liquid phase SP-1000 (layer thickness 0.25 μm). Helium was used as the carrier gas (80 mL/min), the column temperature was 200 °C, and temperatures of the evaporator and detector were 230 and 200 °C, respectively. In addition, 2-methoxyacetophenone was used as the internal standard. PC analysis using NMR was performed on a Bruker AVANCE 600 MHz spectrometer using a WinNMR data processing program from Bruker.
2.4. Calculations
The reaction rate was determined as the maximum gas absorption rate at the inflection point on the graph of the dependence of pressure in the reactor (
p), at time (
t), using the TableCurve 2D program v5.01.05 (by SYSTAT Software Inc. as in Reference [
10]). This was based on the best mathematical description of the indicated dependence
and the determination of the first derivative maximum value
.
3. Results
Table 1 shows a comparison of the various catalytic systems activity during the synthesis of propylene carbonate under the conditions of obtaining a
13C-labeled product.
Even though
Table 1 shows the influence of the processing conditions, the nature and composition of the tested catalytic systems on their activity indicators (i.e., turnover number (TON) and turnover frequency (TOF)), as well as the yield of the PC, the nature of the processes taking place over time did not reveal. Therefore, to develop the most reasonable choice for the catalytic system, the reaction was carried out while automatically recording the pressure and temperature in the reactor in 1-min increments.
Figure 1 shows the dependence of pressure and temperature on time in the presence of various catalytic systems. Despite the different catalytic activities of these systems (see the TON and TOF values in
Table 1), the common feature between them was the pressure drop that began even during the heating of the reaction mass (10–20 min after the start of heating). The reaction completion almost lasted for an hour, which was accompanied by the complete absorption of CO
2 and volatile propylene oxide with a yield of high boiling propylene carbonate near 100%, which was not unusual.
It should be noted that the ZnBr
2/Bu
4N
+Br
− system was used to develop the industrial process of producing
13C-propylene carbonate [
7].
Figure 1 shows the typical curves of the pressure and temperature changes over time for the chosen processing mode of this system.
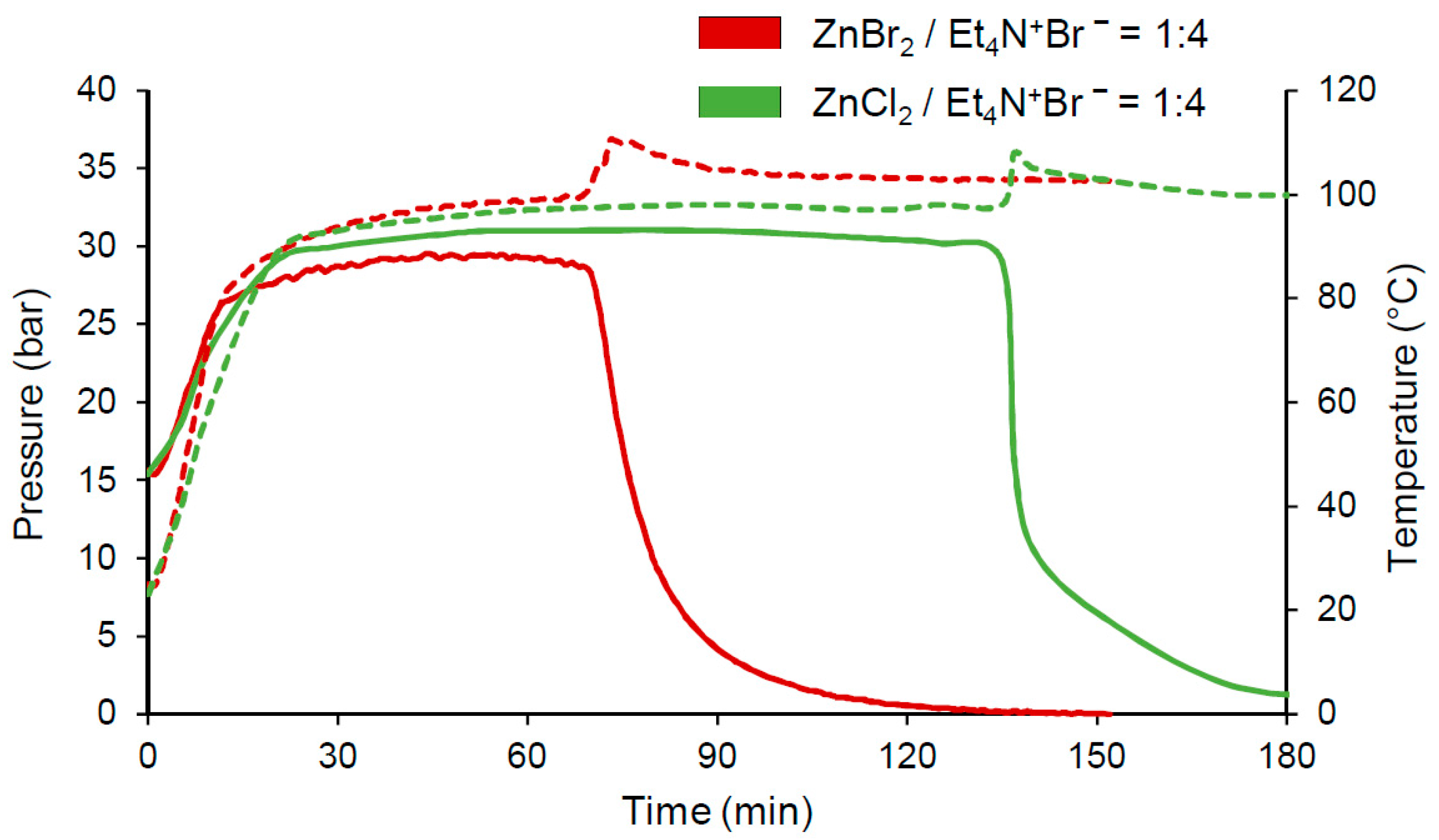
However, using the ZnBr
2/Et
4N
+Br
− catalytic system under the same conditions, these dependencies acquired completely different characteristics (
Figure 2). These include the appearance of a long induction period (over 60 min), followed by rapid gas absorption, and the release of a noticeable amount of heat (peak on the temperature curve). In this case, the reaction was completed at about the same time, starting from the moment the pressure drop began. Under comparable conditions, the absorption rates of the gases (see the experiment section) for the catalytic systems ZnBr
2/Bu
4N
+Br
− and ZnBr
2/Et
4N
+Br
− were 3.5 and 4.4 bar/min, respectively.
Moreover, the features of the kinetics process in the system with Et
4N
+Br
− appeared more clearly when using ZnCl
2 (instead of zinc bromide) (
Figure 2). Under comparable conditions, the induction period increased significantly (from 70 to 135 min) and the gas absorption rate increased by about three times (from 2.5 to 7.6 bar/min). Thus, by using Et
4N
+Br
− in the considered catalytic system based on ZnX
2 (X–halogen), the process ran significantly different to that in the presence of Bu
4N
+Br
−. Interestingly, the maximum TOF value for the ZnCl
2/Et
4N
+Br
− system was 21,658 h
−1 in terms of the highest gas absorption rate (
Figure 2, Run 11 in
Table 1). This was quite large since the greatest activity of the known catalytic system of this process based on porphyrin metal complexes is 46,000 h
−1 [
11].
In the case of the ZnBr
2/Et
4N
+Br
− system, a change in the initial loading of CO
2 and temperature significantly affected the duration of the induction period and the rate of the process.
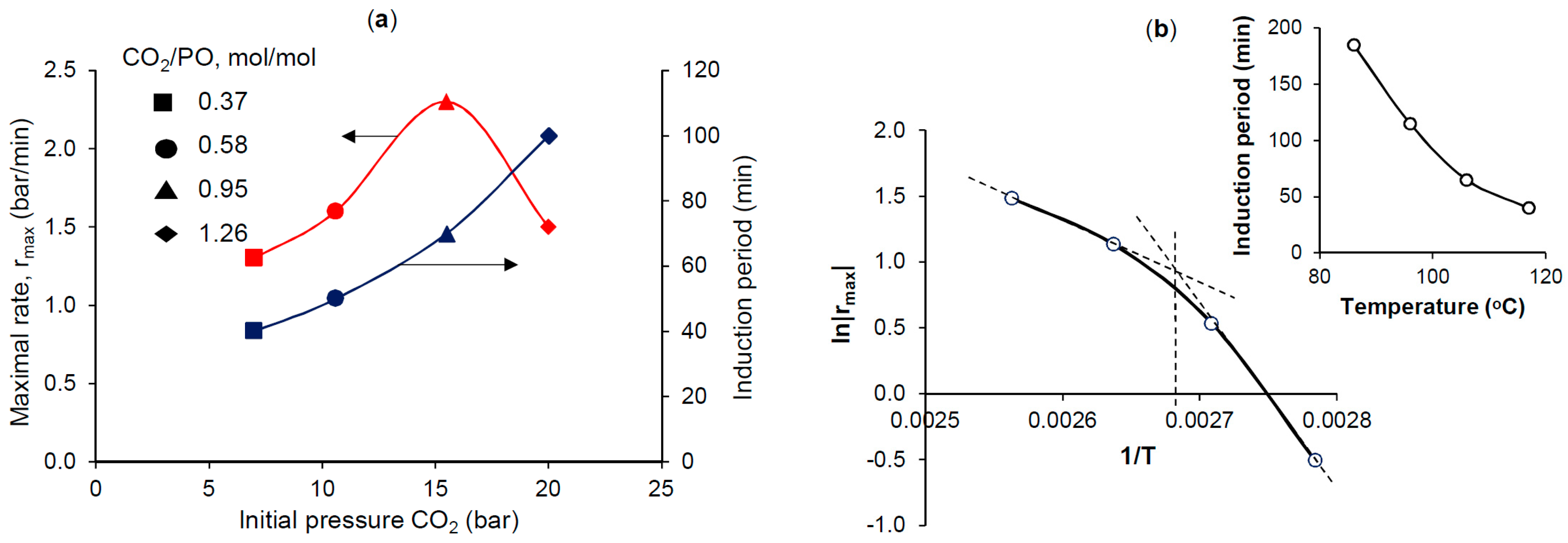
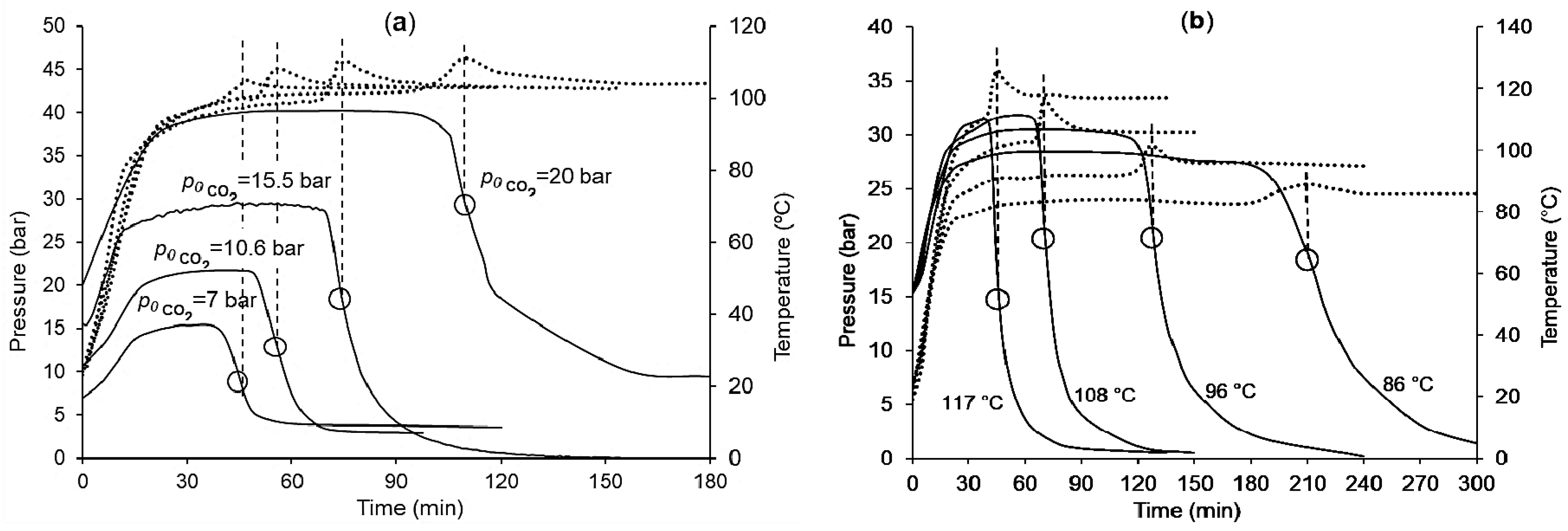
Figure 3a shows that with an increasing
p0 CO
2 from 7 bar to 20 bar, the duration of the induction period is increased 2.5 times reaching about 100 min. At the same time, the process rate varied according to the extremal curve reaching a maximum in the region of the stoichiometric ratio CO
2/PO ≈ 1:1. A temperature increase (
Figure 3b) from 86 °C to 117 °C caused a significant decrease in the induction period (from 185 to 40 min); whilst the temperature dependence of the process rate did not obey the Arrhenius law, which may have indicated a change in the rate-limiting stage at a temperature near 100 °C. Plots of these experiments (dependences of pressure and temperature versus time) are shown in
Figure A1.
To clarify the nature of the induction period and the possibility of generating the active form of the catalyst, an experiment was conducted involving the addition of the reaction mass, formed in the other run during the gas uptake, into the initial mixture of the reaction. For this purpose, the standard experiment was stopped at the start of gas absorption (i.e., at a 10–15% pressure drop level), and the resulting reaction mass at an amount of 7% was added to the initial mixture of the subsequent experiment, where the experiment was carried out under the same conditions. It turned out that such an additive shortened the induction period by almost three times from 72 to 25 min. It was also found that the loading of the CO
2 into the mixture of the catalyst and PO, heated to a given temperature, led to the full disappearance of the induction period (
Figure 4).
4. Discussion
Figure 4 shows that during the induction period, an active complex of PO with a catalyst is formed. The subsequent addition of CO
2 led to a rapid drop in the total pressure in the reactor with the formation of the desired propylene carbonate. The results obtained and the data on the influence of the initial pressure of CO
2 (
Figure 3a) indicated that the active complex was formed, and apparently without the participation of CO
2. In this case, CO
2 could have a deactivating effect on the initial form of the catalyst, for example, by reversibly binding zinc complexes in a manner like Equation (2) [
12,
13], which causes an increase in the induction period duration with the increase in
p0 CO
2. During the process, the catalytically active centers are released, and their concentration increases and, therefore, the reaction rate increases as well. It should be noted that in all the experiments presented in
Figure 4, the PC yield was near quantitative.
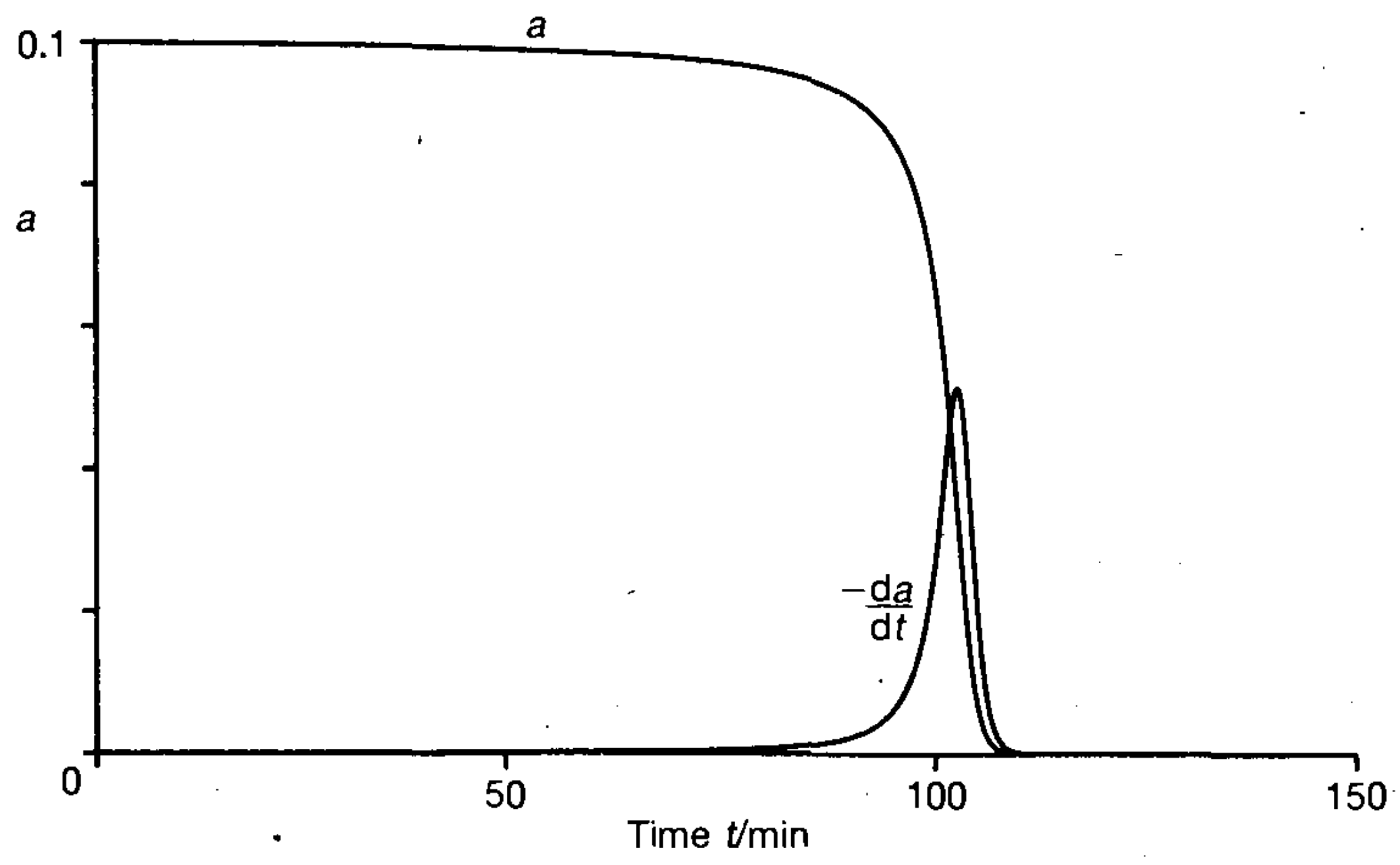
It was noteworthy that in the experiments performed, the induction period reached 70% of the total duration of the synthesis. Such phenomena are characteristic of autocatalytic processes [
14], especially for processes with a “high degree” of autocatalysis and in particular for cubic autocatalysis, for example, the reaction
[
15,
16] (
Figure 5).
Among the known models of autocatalytic processes, a model for the mechanism called the “Brusselator model” described in Reference [
15] (Equation (3)) corresponds very closely to this case:
where A, B, and P mean PO, CO
2, and PC, respectively, while X, Y, and C are the intermediate products of the interaction between PO and CO
2 with a catalyst. In this case, the process rate
follows the equation
, i.e., sharp increases in the rate occur as the X concentration increases, then X transforms into the final product P.
To determine the nature of the intermediate products, the various means of α-oxide carboxylation during the induction period were considered. It is known that these processes can proceed with the formation of not only cyclic carbonates, but also polyethers and polyesters, i.e., polyglycols and polycarbonates according to Equation (4) [
17,
18].
In the absence of water (as in the experiments described above), the synthesis of polyglycols in solutions of metal salts or quaternary ammonium leads to the formation of crown ethers [
19]. On the other hand, polycarbonates formed during the carboxylation of α-oxides can be destructed with the formation of monomeric cyclic carbonates (the “backbiting” mechanism) according to Equation (5) [
12,
20].
Therefore, it could be expected that during the induction period, some of the PO oligomerization products are formed, which then rapidly turn into a target carbonate. In this case, in Equation (3), X, Y, and C are intermediate structures (or complexes) based on polypropylene glycol, CO2, and polypropylene carbonate, respectively. However, attempts to detect such an oligomeric product for a long induction period (by stopping the synthesis, removal of volatile PO under vacuum, and analysis of the residual mass) have proved to be unsuccessful.
Apparently, in our case, during the formation of an active complex of the catalyst and PO, no significant number of high-molecular products were formed, which could be formed in catalytic amounts (see
Table 1). This hypothesis is supported by the literature on the catalytic activity of metal complexes with crown ethers [
21,
22,
23] and polyglycols [
24] in α-oxide carboxylation with the formation of cyclic carbonates. The catalytic activity of hydroxyl derivatives of polyglycols has also been reported, even in the absence of metals, due to the hydroxyl protons which form a hydrogen bond with oxygen in the initial α-oxide [
25]. In addition, several studies [
2,
26] have suggested the activation of CO
2 under the action of quaternary ammonium or phosphonium salts, including the action of liberated free amines or phosphines [
21,
27]. Therefore, in the processes of α-oxide carboxylation in the presence of ZnX
2/R
4N
+X
−, small amounts of polyglycols with terminal ammonium groups can be formed [
28]. Such products can form active complexes that are capable of activating CO
2 using terminal ammonium groups and can also coordinate zinc (similar to crown ethers [
21,
22]) as a Lewis center for α-oxide activation.
Taking into consideration the above, the induction period may be due to the formation of propylene oxide oligomers of a certain size, which are able to coordinate zinc and which can possibly have terminal ammonium groups. This process can proceed together with the gradual release of zinc centers initially deactivated by CO
2 (see the dependence of the induction period on
p0 CO
2). Having reached a required concentration, such complexes catalyze (possibly at a high rate) the process of propylene oxide carboxylation to produce propylene carbonate. In addition, a certain contribution to the formation of the active forms of the catalyst during the induction period can be achieved through the depolymerization of the initial ZnX
2 salt and the formation of complexes, for example, the composition ZnX
2(R
4N
+X
−)
2 [
29]. The duration of this process may depend on the nature of the halogen and the quaternary ammonium salt.
5. Conclusions
Therefore, unlike many well-known catalytic systems for producing propylene carbonate through propylene oxide carboxylation (including metal salts or organic catalysts in the presence of various halides), the use of the ZnX2/Et4N+Br− system (X = Br, Cl) leads to a longer induction period after which the reaction accelerates with the formation of propylene carbonate. In this case, substantial heat release occurs, and very high catalyst productivity is observed (TOF reaches 21,658 h−1 in the region of the maximum process rate values). Under selected conditions (closed system, mole loads of CO2 and PO are comparable), the increase in the initial pressure of the CO2 leads to a noticeable increase in the duration of the induction period. Moreover, at a molar ratio of CO2/PO ≈ 1:1, the rate reaches a maximum and with a further rise in the CO2/PO, the rate decreases. In addition, with an increase in temperature, the induction period is rapidly shortened. The introduction of part of the reaction mass from the gas uptake stage into the initial mixture significantly reduces the induction period which completely disappears when the CO2 is supplied to the heated mixture of the catalyst with PO.
Unfortunately, the data obtained has yet to allow us to understand the reasons for such evident differences in the behavior of ZnBr
2/Et
4N
+Br
− from ZnBr
2/Bu
4N
+Br
−, as well as many other catalytic systems of propylene oxide carboxylation to propylene carbonate [
30]. The clarification of the mechanism requires further study using the method of stoichiometric interaction of propylene oxide with catalytic system components, or by using spectral studies of this reaction in situ. One cannot exclude that regularities like the ones found in this work may be inherent to many other catalytic systems that are used in obtaining cyclic carbonates. Therefore, the study of such features would significantly improve the efficiency of these processes by reducing the induction period while maintaining a high rate of the target product formation using simple and cheap catalytic systems.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}