1. Introduction
The most common materials, used as reinforcement, in polymer resins are: steel, asbestos (fibrous silicate mineral), aramid, and carbon [
1]. Each of them possesses advantages and disadvantages. For example: steel is endowed with an exceptional mechanical strength but also an elevated density. Steel fibers possess a density (7.85 g/cm
3) about three times higher than glass fibers (2.46 g/cm
3), and four times higher than carbon fibers (1.76 g/cm
3) [
2]. Carbon fibers are one of the most expensive fibers, followed by aramid fibers [
3]. The asbestos fibers have good characteristics, as strength, durability, flexibility, and resistance to corrosion, heat, and fire, but are considered harmful given the carcinogenic response in the human body [
4].
Recently, basalt has been proposed as the “twenty-first century non-polluting green material” [
5]. Basalt fibers possess similar performance to glass fibers, and are cheaper compared to carbon fibers [
6]. Although more persistent in biological structures, basalt fibers have displayed a lower acute toxicity and less fibrogenic and carginocenic activity compared to chrysotile asbestos [
7].
Basalt is an aphanitic igneous extrusive (volcanic) rock formed by the rapid cooling of basaltic lava, equivalent to gabbro-norite magma [
8]. Its chemical composition is made prevalently from silicon dioxide (SiO
2, <51.5 wt.%), aluminum oxide (Al
2O
3, <19.0 wt.%), magnesium oxide (MgO, <10.5 wt.%), iron oxide (FeO + Fe
2O
3, <12.0 wt.%), sodium and potassium oxide (K
2O + Na
2O, <6 wt.%), and other remaining species, including titanium oxide (TiO
2), Chromium(III) oxide (Cr
2O
3), manganese oxide (MnO) [
9].
Basalt is hard and dense, directly suitable for fiber manufacturing. Basalt fibers (BF) are produced in a single step process of melting and extruding the raw material at temperatures of 1400–1500 °C and drawing it to form fibers. Contrary to glass fiber (GF) production, no additives (chemicals, solvents, pigments, or hazard materials) are applied during the manufacturing process of basalt fibers [
10].
Due to the growing attention of governments and environmental agencies towards the increasing amount of waste products abandoned in the landfill and polluting disposal operations, several solutions are proposed to recover plastic waste [
11] and shift the production towards more ecofriendly solutions [
12]. The common procedures to reclaim fibers from reinforced composites are mechanical methods, pyrolysis, oxidation in fluidized beds and chemical recycling [
13]. However, thermal incineration remains the most preferred among them: at elevated temperatures (450–600 °C), the polymer matrix is burned off, leaving the residual char and the fibers [
14]. Following incineration, the basalt powder can be employed once again, as a reinforcement, in new composite formulations [
13]. The recycling of basalt fiber can be done in much better environmentally friendly ways than glass fibers [
15]. In fact, during the incineration of discarded glass fiber-based composites, a great amount of black smoke and bad odors are generated. Often glass fibers are melted and create damage to the implants. Moreover, the decomposition of synthetic fibers is not easy and causes a strong environmental load on the reclamation process [
5].
For these reasons, basalt fibers are also considered green, sustainable, natural materials that do not create environmental problems, pose pollution issues, and produce less risks for the health and safety of humans [
16].
Basalt-based composites have been proposed for application in transportation infrastructure [
17], in civil engineering [
18], in marine structures [
19], in automotive [
13], and more recently in armors for ballistic protection [
20], in wind turbines [
21], in clean energy and power grids [
16], in thermal insulation [
22], and in offshore industry [
23].
However, until now, the effect of basalt fibers to improve the mechanical and thermal properties of polymer matrices has not been fully investigated [
24].
Polyamide (PA) is the first synthetic thermoplastic semicrystalline polymer, well-known by the name of Nylon. Its chemical structure is made up of an amide group (-CONH-) as a regular repeating portion of the macromolecular chain. The excellent mechanical strength, the temperature-resistant properties, the high resistance to friction, scratching and solvents, and the noise absorption performances allow the use of polyamide in applications to replace metals, and to satisfy the demand of engineering plastics in electronic, electrical and automobile industries, in household appliances, and sporting goods [
25].
Depending upon the monomer used during synthesis, different types of polyamides (PA 6, PA 66, PA 11, PA 12, PA 6,12) can be realized. PA 66 is an important member of the PA family, often applied in automobiles, textiles, and decoration sectors, characterized by a high stiffness and mechanical strength over a wide temperature range [
26], and a short-term thermal resistance linked to the high melting point [
27]. The repeating unit of PA 66 is constituted of two monomers: the first is the adipic acid, the second is hexamethylene diamine. Each monomer contains six carbon atoms and carries amino and carboxylic acid as functional groups [
28]. Mineral fillers or several fibers are considered the common reinforcing agents in composite materials since they lead to an improvement of the mechanical properties of polymer matrix, and at the same time to a reduction in the cost. The typical mineral filler introduced in PA 66 resin is glass fiber: the resulting PA66/GF composites are mostly applied for the manufacture of high strength parts, especially in the automotive and industries provision of sports and leisure materials [
27].
The non-isothermal crystallization of the polyamide 66 matrix and its composites with glass fiber has been investigated by DSC in the study of Makhlouf et al. [
27]. The crystallization temperature of PA 66/GF composites was found to be up to 7 °C higher than that of the neat matrix.
The thermal behavior of nylon-66 and GF/nylon-66 at 30 wt.% of fiber content was examined by thermogravimetric analysis (TGA) and its derived thermogram (DTA). This characterization has been considered useful for proper understanding of the material processing and fabrication. Activation energy for the polymer degradation, calculated through two different methods (Murray–White and Coats–Redfern) was higher for GF/PA 66 composites compared with unfilled matrix. This finding was intended as a higher thermal stability due to the fiber introduction in neat PA66 polymer [
29].
In this framework, this study aimed to attest the potential applicability of basal fibers to replace the most commonly used glass fibers in polyamide 66 resin. Attention was focused to identify the initial decomposition temperature, the glass transition temperature, the melting and crystallization point, and the shear viscosity. Final outcomes provided useful information on material processing of GF/PA66 and BF/PA66 composites. Usually, the main phases of material process consist of: melting of solid particles, molding into defined shape, and solidification of final product. The knowledge of the thermal stability indicates the limit at which the material can be considered intact (not decomposed) under the influence of temperature. The glass transition indicates the starting point at which the material could be considered workable. The crystallization point denotes the temperature at which, starting from the melted state and then cooling, the polymer macromolecules arrange themselves to form ordered structures. This phenomenon, linked to the cooling degree of the sample during the material shaping, impacts on the processing time and cost. The melting temperature, i.e., the condition at which the ordered polymer crystalline structure is broken through the suppling of heat, controls the energy requirement during the manufacturing process, more than cooling and solidification. The melt resistance affects the resistance to the screw rotation and drive power. The higher the melt resistance the more difficult the material flows within the channel.
3. Results
3.1. Thermal Stability and Kinetic Parameters
TGA and DTGA curves of pure and filled PA 66 with glass and basalt fibers were presented in
Figure 1.
At a heating rate of 20 °C/min, the TGA curve of pure PA 66 (black square points) showed one step of thermal degradation as a result of random chain scission process. The main organic products were eliminated in the form of hydrocarbons, nitriles and vinyl groups [
32]. The thermal decomposition of polyamide 66 is a complex process involving hydrolysis, decarboxylation, deamination and dehydration, with release of water (H
2O), ammonia (NH
3), carbon dioxide (CO
2), cyclopentanone, adipic acid, hexadiamine, nitrile and olefin [
32].
A similar trend of the mass loss (%) curve against temperature was found in the case of composite systems. Regardless of filler type, for composites including a fiber concentration equal to 15 wt.%, the initial decomposition temperature (~410 °C) became higher with respect to neat PA 66 (390 °C), whereas when the filler concentration rose to 25 wt.% the initial decomposition temperature was decreased to about 380 °C. Then, by comparing compounds at an equal fiber content, the initial decomposition temperature was usually higher, even if only by a few degrees (°C), in composites containing basalt fibers against systems containing glass fibers.
These results were also analogously verified by the derivative weight curves (DTGA). The maximum degradation temperature of neat matrix (~441 °C) was shifted to higher values (~454 °C) when the fiber content was equal to 15 wt.% and was moved to smaller values (~425 °C) when the basalt fiber in the matrix amounted to 25 wt.%.
Usually, when fillers have been introduced to a matrix, an increased thermal stability has been verified. This outcome was attributed to the formation of tortuous pathways that slowed down the mass diffusion of the degradation products from the bulk of the polymer to the gas phase. In this case, the fibers acted as a protective barrier against the polymeric thermal decomposition. However, this finding was found to be dependent on filler type and its content. If on one side, when the filler loading was low, a barrier effect could be provoked that led to an improvement in the thermal stability; on the other side, at high filler loading, a promoter effect could be induced that encouraged the degradation process [
33].
From the experimental data, it appeared that, at a content of 15% in wt., the fibers dispersed in the matrix exerted a barrier effect that hindered the polymer matrix decomposition. Then, if the fiber concentration was 25 wt.%, the barrier effect was overcome by the promoter effect, and it was determined to be an initial point for decomposition at lower temperatures with respect to the basic PA 66.
The final residue of PA 66 degradation was evaluated around 1.6% (remaining mass in correspondence of 700 °C). This value increased in composite systems in a proportional way to the introduced nominal filler content in PA 66 polymer during compounding. An appreciable agreement between the effective and the nominal filler content was confirmed.
TGA results were summarized in
Table 1 for all the investigated materials.
The rearranging of TGA data, in terms of weight loss (%) vs temperature (°C), has also made possible the evaluation of kinetic parameters of thermal decomposition for each type of material.
The conversional fraction (α), shown in
Figure 2, was evaluated according to the Equation (2):
where
is the initial sample weight, m is the instantaneous sample weight at certain time t,
is the final sample weight.
The conversion factor of PA 66 remained superior to composites incorporating 15 wt.% of filler loading, but inferior compared to composites at 25 wt.% of filler loading. At an equal fiber percentage, the α curve relating to compounds incorporating basalt fibers was almost coincident with that measured for compounds containing glass fibers.
The kinetics of the degradation reaction were performed by the isothermal rate of conversion
and the following equation (Equation (3)):
where K(T) is the constant of decomposition rate and
is the reaction mechanism function for describing the kinetic thermal degradation [
34].
K(T) is described by the Arrhenius equation (Equation (4)):
where A is the pre-exponential factor,
is the activation energy, and R is the universal gas constant (8.314 J mol
−1 K
−1), T is the temperature in K.
For a heating rate
, the Equation (5) can be written in an integral version (Equation (6)):
f(α) or g(α), defined through several reaction models, were listed in [
35].
Here, it has been considered:
where n is the order of reaction.
The activation energy () has been calculated through the so-called Coats–Redfern method from a plot of ln[G(α)/T2] versus 1/T, at a given conversion, and an order of reaction equal to 1.
The slope of the straight line fitting the experimental data was equal to −
/R, and the corresponding intercept was equal to
[
36].
Figure 3 displayed a comparison among the ln[G(α)/T
2] versus 1/T curves for all the materials.
The almost linear trend of ln[G(α)/T
2] versus 1/T curves with high correlation coefficient (the lowest R
2 = 0.968 for PA6,6) confirmed that a correct model has been used for describing the reaction [
35]. The kinetic parameters, i.e., the activation energy (E
a) and pre-exponential factor (A), were extracted from the
Figure 3 and reported in
Table 2.
Activation energy represents a barrier to be overcome to give life to a chemical reaction. The higher the activation energy, the more difficult it is for the reaction to occur [
36].
The values of Ea were mostly affected by filler content more than filler type. No strong effect of filler type on activation energy of PA 66 was fully demonstrated. The compounds at 25 wt.% of fiber content possessed an activation energy comparable to neat matrix (~170 kJ/mol) that was inferior with respect to composites at 15 wt.% of fiber concentration (~230 kJ/mol). In terms of activation energy, a higher value was found in the case of composites incorporating basalt fibers with respect to those incorporating glass fibers, especially when the fiber content was around 15 wt.%.
E
a here evaluated for neat PA 66 was slightly higher, but comparable, with the values reported in literature of 80–114 kJ/mol [
32,
37,
38] (in all cases heating rate was 10 °C/min). Azimi and Abedifard found an activation energy of 200 kJ/mol, for PA 66 composites containing 10 wt.% of GF [
38].
3.2. Glass Transition, Melt and Crystallization
The DSC thermograms of reinforced PA 66 based composites for heating and cooling treatments are presented in
Figure 4.
First of all, it should be noted that, the properties found during the first heating scan were quite different to those found in the second heating scan for all the investigated materials, particularly for the composite systems. This was intended as a strong influence of the manufacturing process on the thermal characteristics of the compounds. In detail, the addition of both GF and BF in PA 66 matrix led to an increase in the melting temperature. During the first heating (
Figure 4a,b), the melting peak started from a value of 245 °C for neat PA 66 and achieved a value around 256 °C for composites, regardless of filler content and type. The increment of melting temperature in composites both containing glass and basalt fiber, was confirmed also during the second heating (
Figure 4e,f). In this case, the presence of two melting peaks were detected at 245 °C (T
m1) and 251 °C (T
m2) for GF/PA6,6 and BF/PA6,6 composites. The double endothermic peak in the heat flow curve was attributed to the existence of two crystalline structures: the α-crystalline portion and thermodynamically unstable ɣ-crystalline portion [
39].
The crystallization temperature of GF/PA 66 and BF/PA 66 compounds was shifted towards higher temperatures compared to the neat resin. However, the incorporation of glass and basalt fibers into PA 66 matrix also resulted in a decrement in the degree of crystallinity (x
c), evident both in the first and second heating scan (
Table 3).
If on one side, the fibers could act as a nucleating agent by promoting the nucleation of the matrix and increasing the crystallization temperature; then on the other side, when the content was elevated, they could limit the expansion of spherulites by reducing the crystallinity degree [
40]. The reduction in the degree of crystallinity in composites compared to neat matrix, approximately amounted to 30% in the second heating scan, was intended as a strong change in the microstructure of the neat polymer (PA 66) when the fibers have been added. The presence of fibers inhibited the crystallization process, and hence, led to a reduction in the crystallinity degree [
41].
By taking into account the second heating scan, the glass transition temperatures of composites were always comparable among themselves, and about 8 °C lower (~56 °C) than that recorded for the neat matrix (64 °C). Thus, both the glass and basalt fibers added to the PA6,6 have caused an increased chain mobility and lowered the activation energy barrier for segmental relaxations of the surrounding polymer molecules. This effect was attributed to a change in the local segmental package of polymer chains that led to an increase in free volume, and consequently, to the macromolecules mobility [
42].
Finally, it could be concluded that, even if the fiber introduction into PA 66 has strongly affected the melting point, the crystallization and glass transition of the neat polymer, no effect of filler type and content could be highlighted on the thermal characteristics of BF/PA66 and GF/PA66 composites performed through DSC.
The calorimetric results in terms of melting (T
m) and crystallization (T
c) temperatures, melting (ΔH
f) and crystallization (ΔH
c) enthalpies, and the degree of crystallinity (x
c) are reported in
Table 3 for all investigated materials.
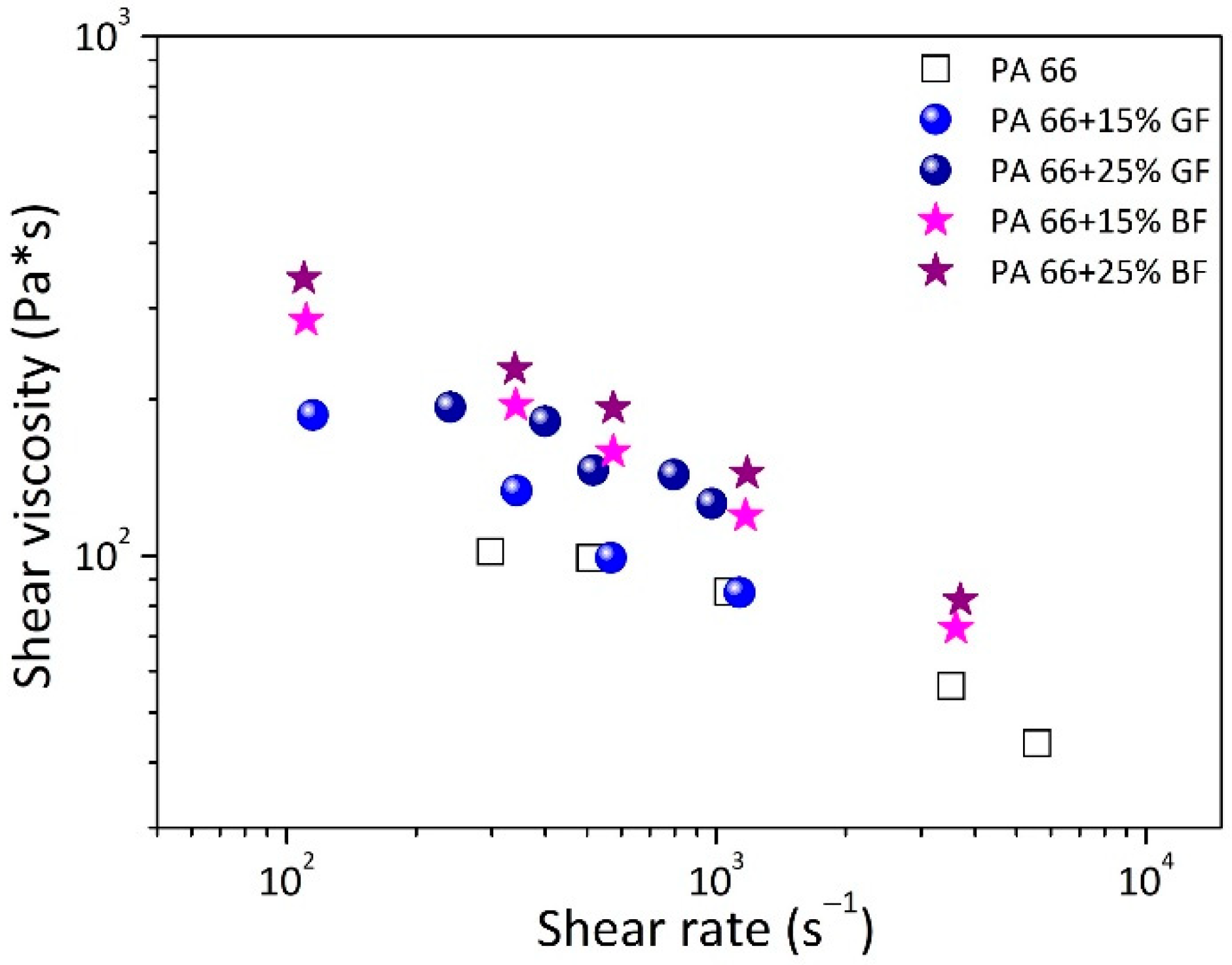
3.3. Shear Viscosity
The processing and shaping of materials are aspects strictly related to their ability to be deformed and to their flow resistance. On this basis, the analysis of rheological properties, developed in a capillary circular die, at temperatures higher than the material melting, and at the typical shear rates encountered in extrusion and injection molding technologies (102–104 s−1), was a practice extremely useful to gain information on the processing behavior of the developed compounds.
The shear viscosity (Pa*s) as a function of the shear rate (s
−1), measured at temperature equal to 270 °C, is displayed in
Figure 5 for GF/PA66 and BF/PA66 composites, and the neat polyamide resin.
Usually, the trend of viscosity vs shear rate for neat polymer can be divided into two regions. The first is characterized by a horizontal stoke, during which the viscosity remained at a constant value, also identified as the Newtonian region. The second is described by a decreasing trend of viscosity as the shear rate was increased, also identified as shear-thinning region or pseudo-plastic behavior. From the data, the viscosity trend of neat PA 66 showed a slightly perceptible Newtonian plateau around a value of 102 Pa*s. After a shear rate of 103 s−1, a shear-thinning trend was followed with decreasing viscosity as the shear rate increased. The flow behavior of neat matrix was fully changed by the introduction of both BF and GF fibers. In fact, in the composites, the Newtonian region was lost and the shear thinning behavior persisted throughout the entire shear rate. The viscosity increase was affected both by filler content and type. At 15 wt.% GF fibers, and higher shear rates (>5 × 102 s−1) the viscosity values were very similar to the PA 66 matrix and detached from those of the neat resin as the shear rate decreased. At 25 wt.% GF content, the shear viscosity was found to be over the values found for composites at 15 wt.% of GF loadings, but slightly lower compared to composites at 15 wt.% BF fibers. Finally, at 25 wt.% BF content, the highest increase in melt viscosity was recorded compared to all the other investigated systems.
It can be supposed that the introduction of fibers in PA 66 led to a constraint in the macromolecular chains’ motion that increased the viscosity of the melted systems compared to the basic polymer, particularly at low shear rate. It was demonstrated that at low shear rates, the fibers were moved and rotated with respect to the flow path, assuming a random position. This led to a greater possibility of fiber-to-fiber collision which resulted in an increased flow resistance, also as the fiber content was increased. At high shear rate the material moved almost like a plug, the fibers became oriented in the flow direction, the fiber–fiber collision was much less possible, and the viscosity increase, due to the increase in content of fibers present, was lower [
43]. Furthermore, two other factors could contribute to the mild increase in viscosity of composites compared to the matrix at high shear rates: the first was the possible breakdown of the fibers at the capillary inlet, which were thus more easily dispersed and oriented in the flow direction; the second was the weak interaction between fiber and matrix due to poor compatibility between the two phases [
33].
The simplest model that can be used to capture such behavior is the power-law, or Ostwald-de Waele model [
44]:
where
is the shear rate, k is the consistency (Pa*s
n) and n is the flow index. Shear thinning behavior is represented by n < 1; shear-thickening behavior is symbolized by n > 1; Newtonian behavior is associated to n = 1. For n in the range between 0 and 1, the smaller the n value, the greater the degree of shear-thinning and the sensitivity of polymer to the shear rate changes (beneficial for the processing).
The experimental data reported in
Figure 5 was fitted according to the power law equation (Equation (9)). The consistency and flow index were evaluated, and reported in
Figure 6, both for matrix and corresponding GF/PA66 and BF/PA66 composites. The coefficients of determination (R
2) resulted to be superior to 0.98 for any regression.
It can be observed that consistency strongly increased with the filler content, and particularly in the case of basalt fibers. For BF/PA 66 composites at 25 wt.% of loading, the consistency grew by four-fold with respect to the value evaluated for basic PA 66. On the contrary, the flow index showed a very weak dependency on the filler concentration, particularly in the case of glass fibers. Actually, the value was equal to 0.7 for the neat polymer and oscillated around 0.6 in the case of composites. In other words, the shear thinning degree of composites at 15 and 25 wt.% GF concentrations, was very similar to the matrix, whereas in the case of composites containing BF, the shear thinning attitude seemed to slightly increase by increasing the fiber loadings. In this latter case, the higher degree of shear thinning was explained with an increase in local shear rate induced in the thin polymer layer in the space between the BF fibers [
45].
3.4. Morphological Aspects
SEM images of GF/PA 66 compounds are reported in
Figure 7a,b at 15 and 25 wt.% in fiber content, respectively. BF/PA66 is shown in
Figure 7d,e, at 15 and 25 wt.%, in fiber content, respectively.
In both systems, a random orientation of fibers throughout the matrix was observed. At 15 wt.% of filler loading, the fibers appeared well isolated from each other in the polymer resin, without evident signs of agglomeration. There were different considerations in the case of composites at 25 wt.% of fiber. In detail, in the GF/PA66 composites, undispersed fiber bundles could be observed. In the case of BF/PA66 composites, the fibers seemed to be more than double those contained in the sample at loading equal to 15 wt.%. These last observations attested to a worsening of fiber dispersion when the related content was increased in the matrix.
However, the presence of cavities and holes on the samples’ surface was considered a direct consequence of pull out and debonding phenomena occurring during the preparation of cryogenic fracture surfaces. These phenomena were usually verified in the field of reinforced polymer matrices. Under load conditions, at a certain point the matrix began to yield. The fiber continued to sustain the load up to a limit that corresponded to the failure strength of filler/matrix interface (debonding). Beyond this limit, the fiber was deformed until it slipped out, leaving a vacuum in the matrix (pull-out) [
33].
The extracted fibers showed no signs of polymer wrapping and surface covered by polyamide (
Figure 7c,f). This indicated that the fibers and matrix were not well combined, and weak interfacial adhesion between the two phases was achieved both in the case of GF and BF compounds.
4. Conclusions
In this work, the performance of BF/PA66 and GF/PA66 composites (at fiber contents of 15 and 25 wt.%) have been compared in terms of thermal stability, glass transition, crystallization and melting point, shear viscosity, and morphological aspects.
Depending upon the content, the presence of added fibers (both BF and GF) to PA 66 matrix could speed up or slow down the polymer decomposition by heat. PA66 matrix possessed a thermal stability lower than composites at 15 wt.% of fiber loading, and higher than systems incorporating 25 wt.% fiber content. The initial decomposition temperature and evaluated activation energy for thermal degradation, were found to be higher in the case of composites incorporating basalt fibers, particularly at 15 wt.% in content, albeit mildly.
The crystallization temperature of BF/PA66 and GF/PA66 systems was determined to be around 218 °C, that was about 30 °C higher with respect to the value of basic PA66. On the contrary, the degree of crystallinity was decreased approximately 20% when the fibers were incorporated into the neat polymer. The presence of fibers, acting as nucleating agents, promoted the crystallization during the cooling phase of the polymeric melt, but the formation of crystalline structures was limited by the large dimensions of the fibers themselves (reducing the crystallinity degree). As concerning the melting point, the existence of a double endothermic peak at 245 °C and 251 °C for the BF/PA66 and GF/PA66 systems, instead of a single endothermic peak at 243 °C displayed for the neat matrix, was attributed to the formation of two crystalline arrangements, one stable and another thermodynamically unstable. A similar glass transition temperature (equal to 56 °C) was verified for all the composites, however they were approximately 7–8 °C lower than the neat matrix (~64 °C).
The melt viscosity of the neat PA66 displayed a perceptible Newtonian plateau followed by a decreasing trend, increasing the shear rate. In the case of composites, the Newtonian plateau disappeared, and a shear thinning behavior was observed with viscosity values higher than the neat resin, particularly at low shear rate and in the BF/PA66 systems. Morphological aspects from SEM micrographs revealed debonding and pull-out phenomena of fibers from the matrix, leaving holes on the sample surface and bare pieces of fiber without polymer covering. These aspects were considered signs of poor compatibility between filler and matrix, both in the case of BF/PA66 and GF/PA66 composites.
Finally, it was concluded that the effect of basalt content introduced into the PA 66 matrix equal to 15 or 25 wt.% on investigated features was quite similar, and comparable to that caused by glass fiber incorporation, at the same amount, in the same matrix. A future study could aim to improve the compatibility and interfacial adhesion between the two phases by proposing appropriate formulations involving fiber sizing, coupling, and/or agents to enhance compatibility.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}