
Enhanced Oxygen Vacancy Formation in CeO2-Based Materials and the Water–Gas Shift Performance
Abstract
:1. Introduction
2. Experimental Procedure
2.1. Preparation of Catalysts
2.2. Catalyst Characterization
2.3. Water–Gas Shift Activity
3. Results and Discussion
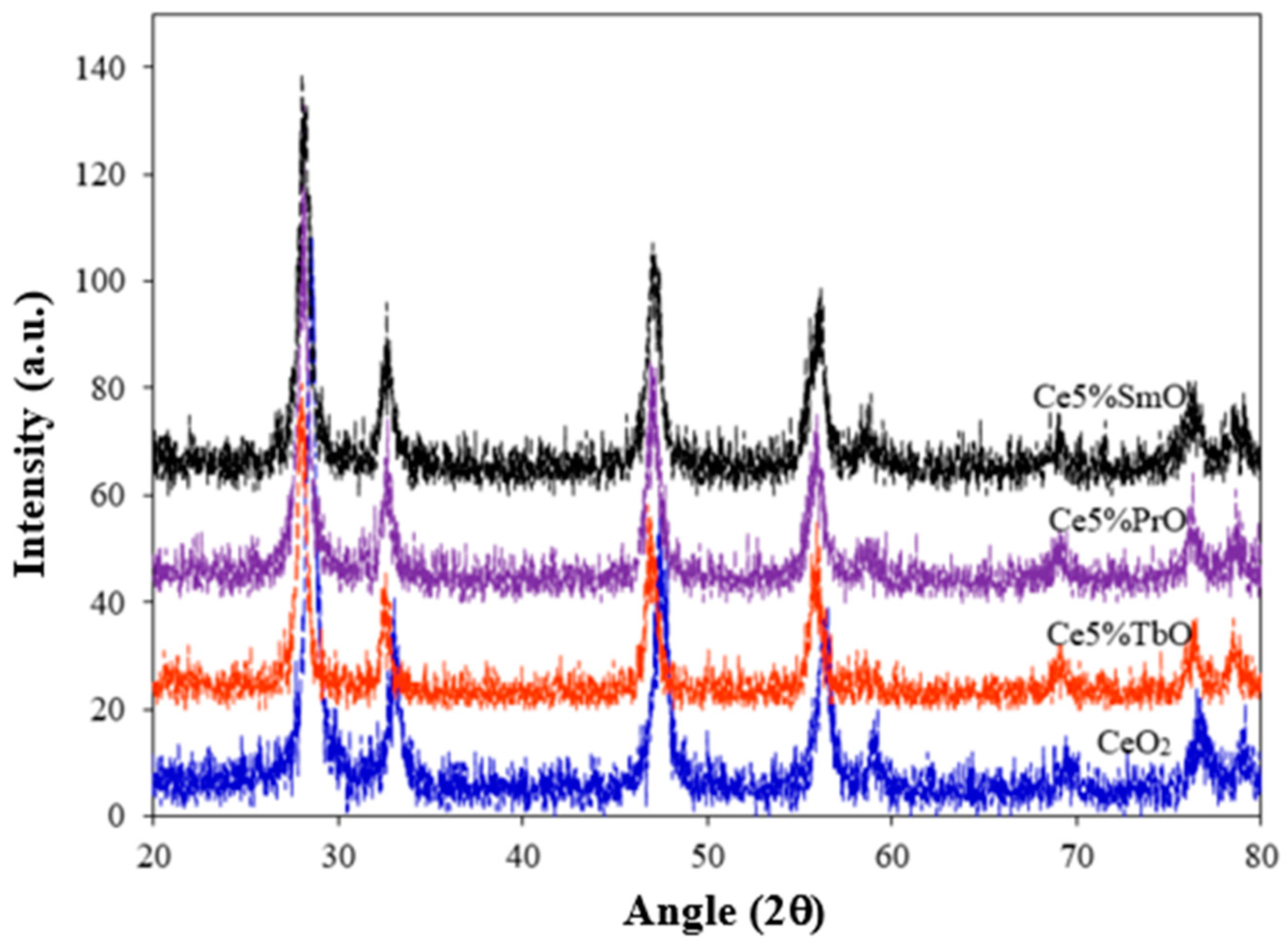
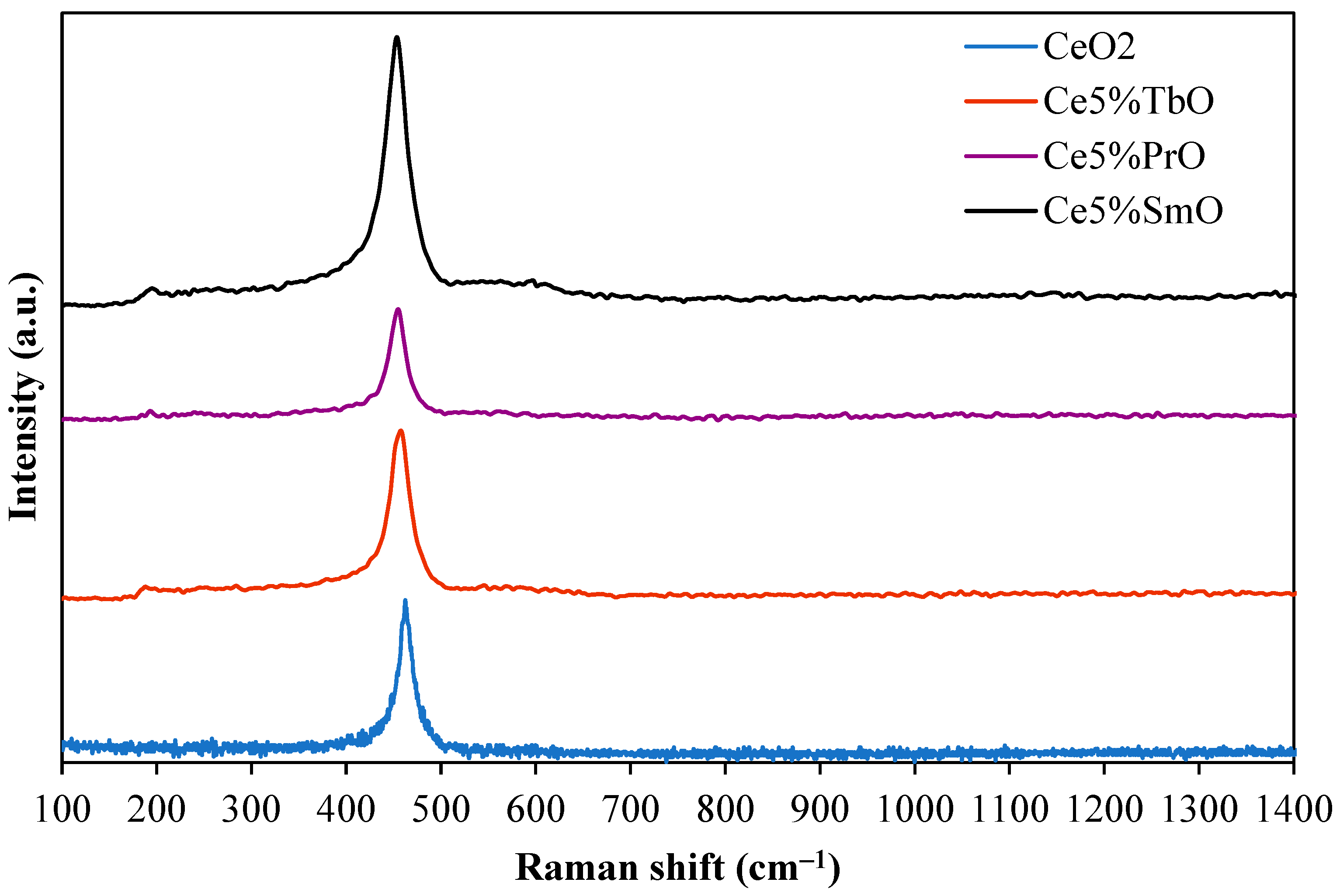
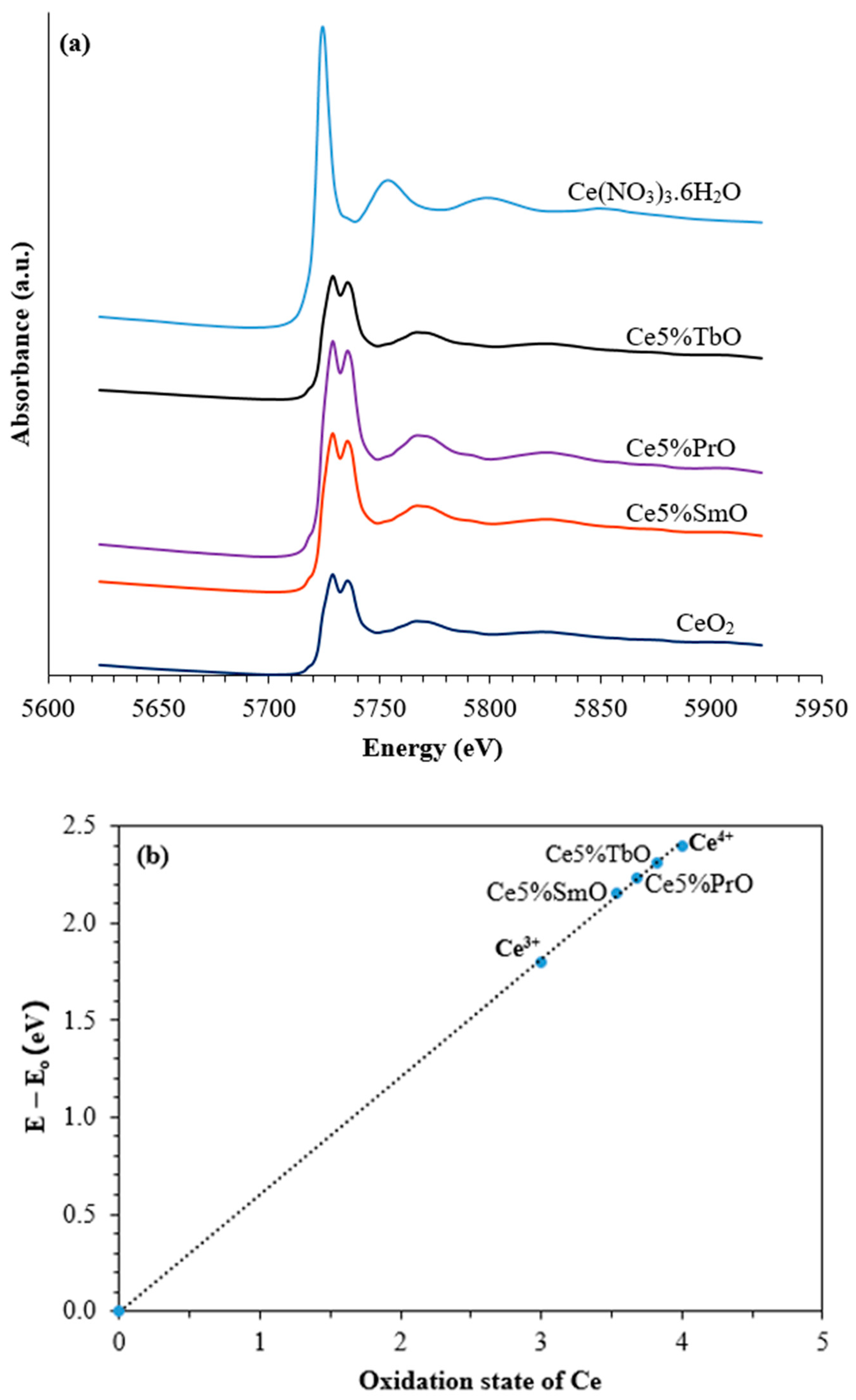
3.1. Characterization of Catalysts
3.2. Water–Gas Shift Performance
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mendes, D.; Mendes, A.; Madeira, L.M.; Iulianelli, A.; Sousa, J.M.; Basile, A. The water-gas shift reaction: From conventional catalytic systems to Pd-based membrane reactors—A review. Asia-Pac. J. Chem. Eng. 2010, 5, 111–137. [Google Scholar] [CrossRef]
- Zhou, F.; Andreasen, S.J.; Kær, S.K.; Park, J.O. Experimental investigation of carbon monoxide poisoning effect on a PBI/H3PO4 high temperature polymer electrolyte membrane fuel cell: Influence of anode humidification and carbon dioxide. Int. J. Hydrogen Energy 2015, 40, 14932–14941. [Google Scholar] [CrossRef]
- Gokhale, A.A.; Dumesic, J.A.; Mavrikakis, M. On the Mechanism of Low-Temperature Water Gas Shift Reaction on Copper. J. Am. Chem. Soc. 2008, 130, 1402–1414. [Google Scholar] [CrossRef]
- Grabow, L.C.; Gokhale, A.A.; Evans, S.T.; Dumesic, J.A.; Mavrikakis, M. Mechanism of the water gas shift reaction on Pt: First principles, experiments, and microkinetic modeling. J. Phys. Chem. C 2008, 112, 4608–4617. [Google Scholar] [CrossRef]
- Lomonaco, J.G.; Sesuk, T.; Charojrochkul, S.; Tepamatr, P. Effect of Re addition on the water–gas shift activity of Ni catalyst supported by mixed oxide materials for H2 production. Catalysts 2023, 13, 959. [Google Scholar] [CrossRef]
- Tojira, O.; Tepamatr, P. Catalytic activity of Ni based materials prepared by different methods for hydrogen production via the water gas shift reaction. Catalysts 2023, 13, 176. [Google Scholar] [CrossRef]
- Tepamatr, P.; Buarod, E.; Laosiripojana, N.; Charojrochkul, S. Study of water gas shift reaction over ceria based catalysts in solid oxide fuel cells. ECS Trans. 2015, 68, 1207–1217. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Papavasiliou, J.; Avgouropoulos, G.; Ioannides, T.; Kondarides, D.I. Water–gas shift activity of doped Pt/CeO2 catalysts. Chem. Eng. J. 2007, 134, 16–22. [Google Scholar] [CrossRef]
- Sadykov, V.A.; Kuznetsova, T.G.; Alikina, G.M.; Frolova, Y.V.; Lukashevich, A.I.; Potapova, Y.V.; Muzykantov, V.S.; Rogov, V.A.; Kriventsov, V.V.; Kochubei, D.I.; et al. Ceria-based fluorite-like oxide solid solutions as catalysts of methane selective oxidation into syngas by the lattice oxygen: Synthesis, characterization and performance. Catal. Today 2004, 93–95, 43–53. [Google Scholar] [CrossRef]
- Tabakova, T.; Idakiev, V.; Papavasiliou, J.; Avgouropoulos, G.; Ioannides, T. Effect of additives on the WGS activity of combustion synthesized CuO/CeO2 catalysts. Catal. Commun. 2007, 8, 101–106. [Google Scholar] [CrossRef]
- Yang, S.C.; Pang, S.H.; Sulmonetti, T.P.; Su, W.N.; Lee, J.F.; Hwang, B.J.; Jones, C.W. Synergy between ceria oxygen vacancies and Cu nanoparticles facilitates the catalytic conversion of CO2 to CO under mild conditions. ACS Catal. 2018, 8, 12056–12066. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalysis by ceria and related materials. In Catalytic Science Series: Volume 2; World Scientific Publishing: Singapore, 2002. [Google Scholar] [CrossRef]
- Mahamulkar, S.; Yin, K.; Claure, M.T.; Davis, R.J.; Li, L.; Shibata, H.; Malek, A.; Jones, C.W.; Agrawal, P.K. Thermally stable α-alumina supported ceria for coking resistance and oxidation of radical coke generated in-situ. Fuel 2018, 218, 357–365. [Google Scholar] [CrossRef]
- Dai, B.; Cao, S.; Xie, H.; Zhou, G.; Chen, S. Reduction of CO2 to CO via reverse water-gas shift reaction over CeO2 catalyst. Korean J. Chem. Eng. 2017, 35, 421–427. [Google Scholar] [CrossRef]
- Vinodkumar, T.; Rao, B.G.; Reddy, B.M. Influence of isovalent and aliovalent dopants on the reactivity of cerium oxide for catalytic applications. Catal. Today 2015, 253, 57–64. [Google Scholar] [CrossRef]
- Mukherjee, D.; Rao, B.G.; Reddy, B.M. Characterization of ceria–based nano–oxide catalysts by Raman spectroscopy. Top. Catal. 2017, 60, 1673–1681. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Element | Found Metal Content a (wt.%) | BET Surface Area b (m2/g) | Average Pore Diameter b (nm) | Crystallite Size c (nm) | Lattice Parameter c (nm) |
|---|---|---|---|---|---|---|
| CeO2 | 64 | 13.48 | 13.59 | 0.5412 | ||
| Ce5%TbO | Tb | 4.9534 | 68 | 13.32 | 13.55 | 0.5483 |
| Ce5%PrO | Pr | 4.8822 | 64 | 14.12 | 16.13 | 0.5504 |
| Ce5%SmO | Sm | 4.9248 | 95 | 9.54 | 13.43 | 0.5505 |
| Catalysts | H2–TPR Peak Position (°C) | H2 Consumption (mmol/g) |
|---|---|---|
| CeO2 | 506 | 0.88 |
| Ce5%TbO | 510 | 0.77 |
| Ce5%PrO | 480 | 0.90 |
| Ce5%SmO | 366, 475 | 0.92 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaewtong, S.; Sesuk, T.; Tepamatr, P. Enhanced Oxygen Vacancy Formation in CeO2-Based Materials and the Water–Gas Shift Performance. ChemEngineering 2024, 8, 79. https://doi.org/10.3390/chemengineering8040079
Kaewtong S, Sesuk T, Tepamatr P. Enhanced Oxygen Vacancy Formation in CeO2-Based Materials and the Water–Gas Shift Performance. ChemEngineering. 2024; 8(4):79. https://doi.org/10.3390/chemengineering8040079
Chicago/Turabian StyleKaewtong, Sangaroon, Thanathon Sesuk, and Pannipa Tepamatr. 2024. "Enhanced Oxygen Vacancy Formation in CeO2-Based Materials and the Water–Gas Shift Performance" ChemEngineering 8, no. 4: 79. https://doi.org/10.3390/chemengineering8040079