
Fluorometric Detection of SARS-CoV-2 Single-Nucleotide Variant L452R Using Ligation-Based Isothermal Gene Amplification
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Oligonucleotides, Enzymes, and Chemical Reagents
2.2. SNM-Specific Ligation
2.3. SDA Reaction
2.4. Pre-Ligated Dumbbell Padlock DNAs
2.5. GQ-RCA and One-Pot SDA/GQ-RCA with Fluorescence Analysis
3. Results and Discussion
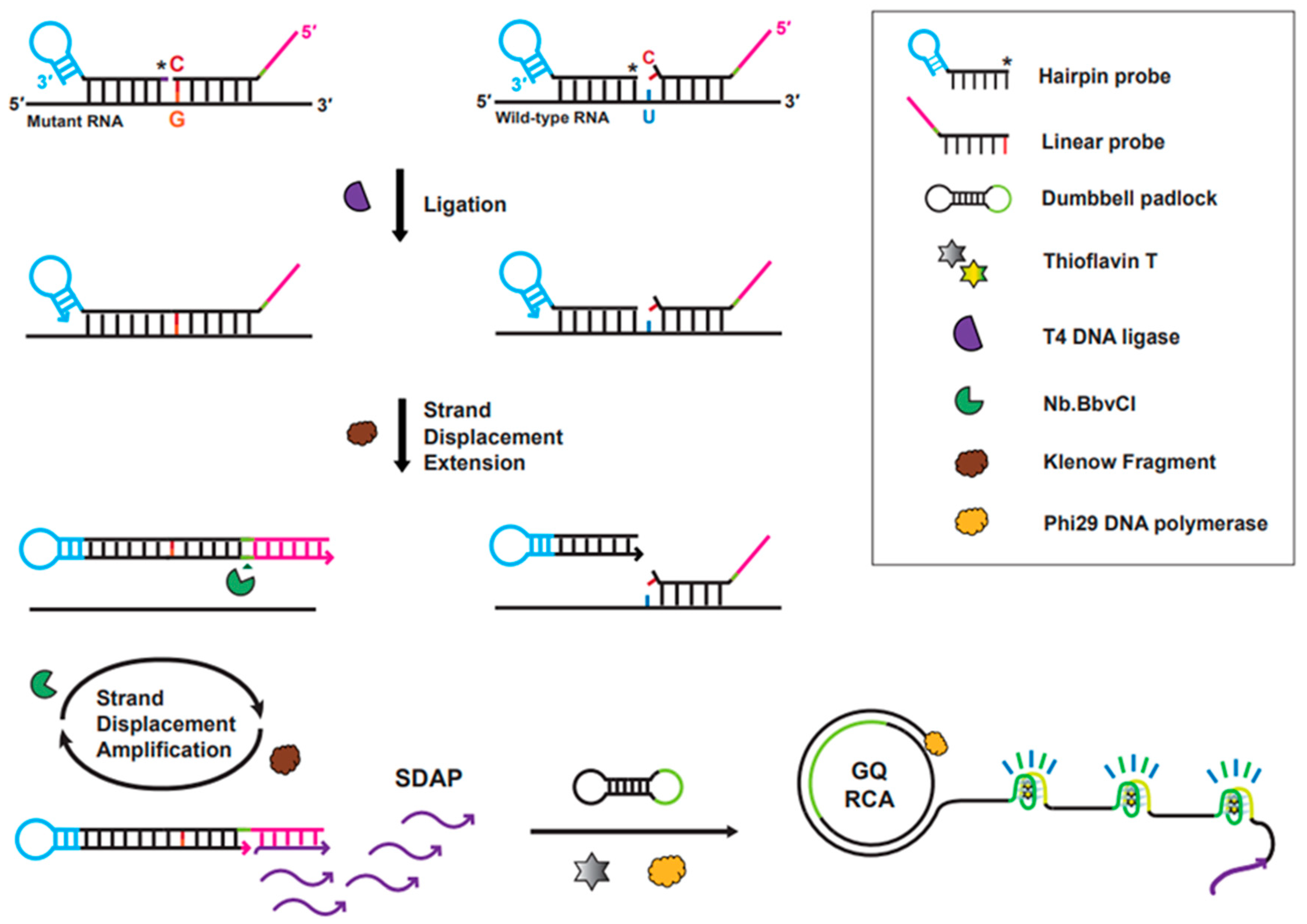
3.1. Strategy of the Ligation-Based Tandem Gene Amplification for Detection of SNM
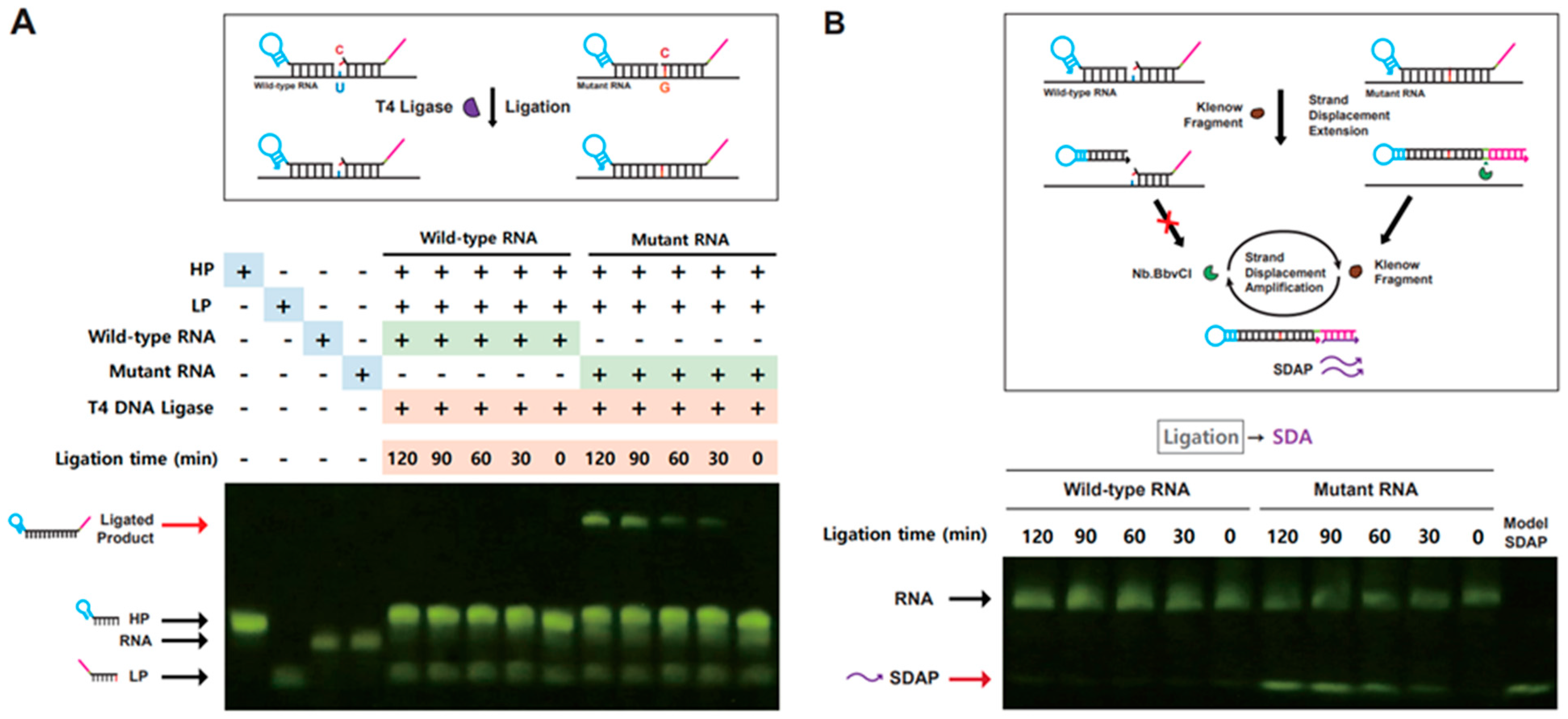
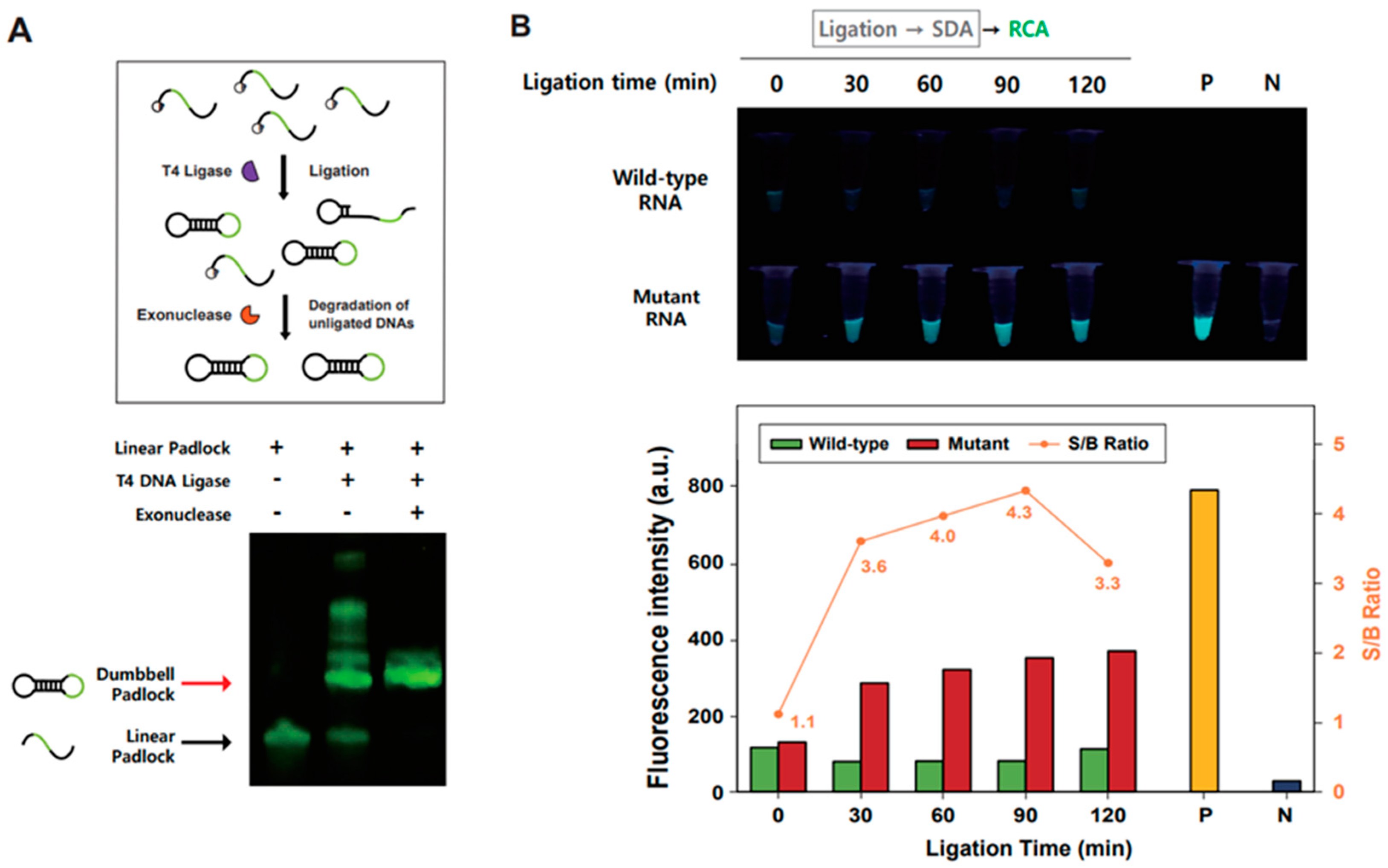
3.2. Validation of Ligation-Based Tandem Gene Amplification for Detection of SNM in RNA
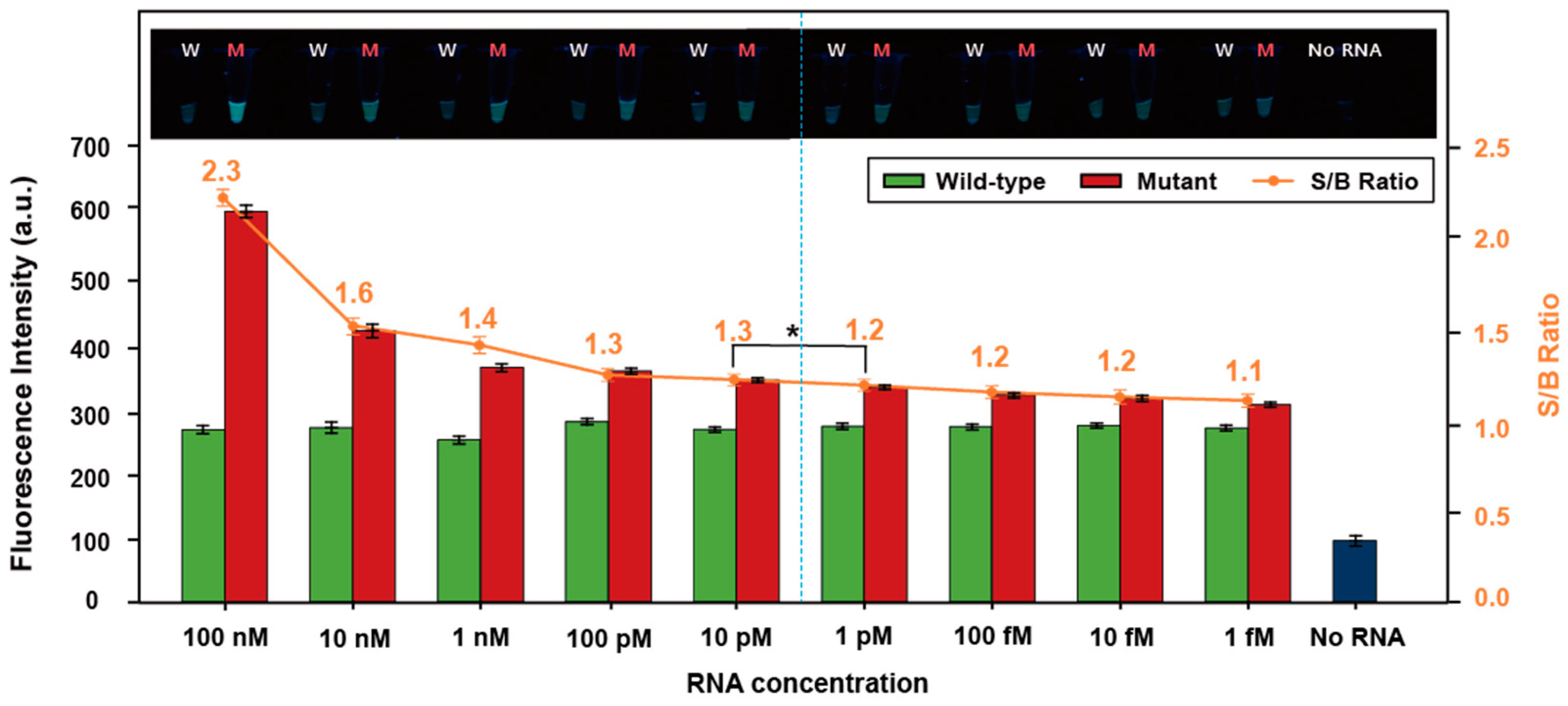
3.3. Sensitivity of the Ligation-Based Tandem Gene Amplification
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Larsen, H.D.; Fonager, J.; Lomholt, F.K.; Dalby, T.; Benedetti, G.; Kristensen, B.; Urth, T.R.; Rasmussen, M.; Lassauniere, R.; Rasmussen, T.B.; et al. Preliminary report of an outbreak of SARS-CoV-2 in mink and mink farmers associated with community spread, Denmark, June to November 2020. Euro Surveill. 2021, 26, 2100009. [Google Scholar] [CrossRef]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136.e1111. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-Gonzalez, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and antibody neutralization of an emerging SARS-CoV-2 variant in California carrying a L452R spike protein mutation. medRxiv 2021, preprint. [Google Scholar] [CrossRef]
- Callaway, E. Making sense of coronavirus mutation. Nature 2020, 585, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Iijima, T.; Ando, S.; Kanamori, D.; Kuroda, K.; Nomura, T.; Tisi, L.; Kilgore, P.E.; Percy, N.; Kohase, H.; Hayakawa, S.; et al. Detection of SARS-CoV-2 and the L452R spike mutation using reverse transcription loop-mediated isothermal amplification plus bioluminescent assay in real-time (RT-LAMP-BART). PLoS ONE 2022, 17, e0265748. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef]
- Jia, Z.; Gong, W. Will Mutations in the Spike Protein of SARS-CoV-2 Lead to the Failure of COVID-19 Vaccines? J. Korean Med. Sci. 2021, 36, e124. [Google Scholar] [CrossRef] [PubMed]
- Tali, S.H.S.; LeBlanc, J.J.; Nikpour, N.; Armanfard, N.; Sadiq, Z.; Sagan, S.M.; Oyewunmi, O.D.; Jahanshahi-Anbuhi, S.; Camargo, C. Tools and Techniques for Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)/COVID-19 Detection. Clin. Microbiol. Rev. 2021, 34, e00228-20. [Google Scholar]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, e63. [Google Scholar] [CrossRef]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA detection using recombination proteins. PLoS Biol. 2006, 4, e204. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.T.; Fraiser, M.S.; Schram, J.L.; Little, M.C.; Nadeau, J.G.; Malinowski, D.P. Strand displacement amplification-an isothermal, in vitro DNA amplification technique. Nucleic Acids Res. 1992, 20, 1691–1696. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.; Li, F.; Zhang, Z.; Zhang, K.; Kang, D.K.; Ankrum, J.A.; Le, X.C.; Zhao, W. Rolling circle amplification: A versatile tool for chemical biology, materials science and medicine. Chem. Soc. Rev. 2014, 43, 3324–3341. [Google Scholar] [CrossRef]
- Walker, G.T.; Little, M.C.; Nadeau, J.G.; Shank, D.D. Isothermal in vitro amplification of DNA by a restriction enzyme/DNA polymerase system. Proc. Nati. Acad. Sci. USA 1992, 89, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Toley, B.J.; Covelli, I.; Belousov, Y.; Ramachandran, S.; Kline, E.; Scarr, N.; Vermeulen, N.; Mahoney, W.; Lutz, B.R.; Yager, P. Isothermal strand displacement amplification (iSDA): A rapid and sensitive method of nucleic acid amplification for point-of-care diagnosis. Analyst 2015, 140, 7540–7549. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Kang, P.J.; Chen, Z.P.; Shi, C.X.; Chen, Y.; Yu, R.Q. Highly specific and sensitive detection of microRNAs by tandem signal amplification based on duplex-specific nuclease and strand displacement. Chem. Commun. 2019, 55, 14210–14213. [Google Scholar] [CrossRef]
- Lee, H.; Kim, D.M.; Kim, D.E. Label-free fluorometric detection of influenza viral RNA by strand displacement coupled with rolling circle amplification. Analyst 2021, 145, 8002–8007. [Google Scholar] [CrossRef]
- Lee, H.; Lee, H.; Hwang, S.H.; Jeong, W.; Kim, D.E. Detection of SARS-CoV-2 RNA through tandem isothermal gene amplification without reverse transcription. Anal. Chim. Acta 2022, 1212, 339909. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.Q. Rolling replication of short DNA circles. Proc. Natl. Acad. Sci. USA 1995, 92, 4641–4645. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Y.; Yang, Y.; Huang, J.; Wang, H.; Zhu, Z.; Wang, X.; Ma, P.; Zhou, X.; Wang, S.; et al. Ligation-promoted hyperbranched rolling circle amplification enables ultrasensitive detection of microRNA in clinical specimens. Sens. Actuators B Chem. 2018, 277, 634–639. [Google Scholar] [CrossRef]
- Mohsen, M.G.; Kool, E.T. The Discovery of Rolling Circle Amplification and Rolling Circle Transcription. Acc. Chem. Res. 2016, 49, 2540–2550. [Google Scholar] [CrossRef]
- Goo, N.I.; Kim, D.E. Rolling circle amplification as isothermal gene amplification in molecular diagnostics. Biochip J. 2016, 10, 262–271. [Google Scholar] [CrossRef]
- Qi, X.; Bakht, S.; Devos, K.M.; Gale, M.D.; Osbourn, A. L-RCA (ligation-rolling circle amplification): A general method for genotyping of single nucleotide polymorphisms (SNPs). Nucleic Acids Res. 2001, 29, e116. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Ohkawachi, M.; Horio, K.; Kobori, T.; Aki, T.; Matsumura, Y.; Nakashimada, Y.; Okamura, Y. RNase H-assisted RNA-primed rolling circle amplification for targeted RNA sequence detection. Sci. Rep. 2018, 8, 7770. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-M.; Seo, J.; Jun, B.-H.; Kim, D.H.; Jeong, W.; Hwang, S.-H.; Kim, D.-E. Fluorometric detection of influenza virus RNA by PCR-coupled rolling circle amplification generating G-quadruplex. Sens. Actuators B Chem. 2017, 251, 894–901. [Google Scholar] [CrossRef]
- Mohanty, J.; Barooah, N.; Dhamodharan, V.; Harikrishna, S.; Pradeepkumar, P.I.; Bhasikuttan, A.C. Thioflavin T as an efficient inducer and selective fluorescent sensor for the human telomeric G-quadruplex DNA. J. Am. Chem. Soc. 2013, 135, 367–376. [Google Scholar] [CrossRef]
- Kim, M.; Kim, D.M.; Kim, D.E. Label-free fluorometric detection of microRNA using isothermal rolling circle amplification generating tandem G-quadruplex. Analyst 2020, 145, 6130–6137. [Google Scholar] [CrossRef]
- Lee, I.J.; Goo, N.I.; Kim, D.E. Label/quencher-free detection of single-nucleotide changes in DNA using isothermal amplification and G-quadruplexes. Analyst 2016, 141, 6503–6506. [Google Scholar] [CrossRef]
- Kim, D.-M.; Seo, J.; Kim, D.-W.; Jeong, W.; Hwang, S.-H.; Kim, D.-E. Fluorometric detection of single-nucleotide mutations using tandem gene amplification. Sens. Actuators B Chem. 2020, 314, 128071. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kyung, K.; Ku, J.; Cho, E.; Ryu, J.; Woo, J.; Jung, W.; Kim, D.-E. Fluorometric Detection of SARS-CoV-2 Single-Nucleotide Variant L452R Using Ligation-Based Isothermal Gene Amplification. Bioengineering 2023, 10, 1116. https://doi.org/10.3390/bioengineering10101116
Kyung K, Ku J, Cho E, Ryu J, Woo J, Jung W, Kim D-E. Fluorometric Detection of SARS-CoV-2 Single-Nucleotide Variant L452R Using Ligation-Based Isothermal Gene Amplification. Bioengineering. 2023; 10(10):1116. https://doi.org/10.3390/bioengineering10101116
Chicago/Turabian StyleKyung, Kangwuk, Jamin Ku, Eunbin Cho, Junhyung Ryu, Jin Woo, Woong Jung, and Dong-Eun Kim. 2023. "Fluorometric Detection of SARS-CoV-2 Single-Nucleotide Variant L452R Using Ligation-Based Isothermal Gene Amplification" Bioengineering 10, no. 10: 1116. https://doi.org/10.3390/bioengineering10101116