

Prospection of Peptide Inhibitors of Thrombin from Diverse Origins Using a Machine Learning Pipeline



Abstract

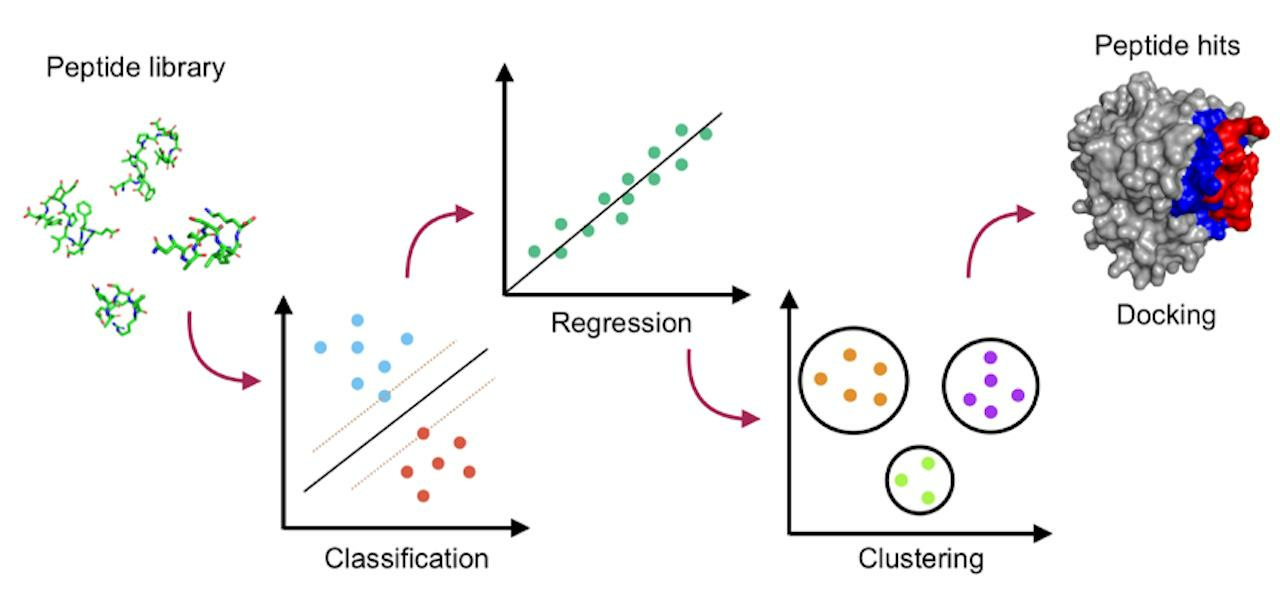
1. Introduction
2. Methods
2.1. Dataset Preparation
- Positive Dataset. We collected only direct thrombin-inhibiting peptides reported as “antithrombotic” from peer-reviewed publications. We collected the sequences of these peptides from UniProt [24], NCBI Protein Database [25], RCSB PDB [26], and PubChem [27]. Next, we obtained the experimentally determined inhibition constants of these peptides against thrombin, also from peer-reviewed publications. After removing the duplicates, we obtained 88 naturally occurring antithrombotic peptides, and inhibition constants of 53 of these peptides (Table S1). Only peptides containing naturally occurring amino acids were chosen.
- Negative Dataset. To prepare the non-antithrombotic negative dataset, we collected peptides from the UniProt and NCBI databases that were not annotated as “anticoagulant”, “antithrombotic”, “hemostasis-impairing”, “antimicrobial”, “anti-inflammatory”, or “thrombin inhibitor”. To minimize bias in the random selection of peptides agnostic to thrombin binding, the ratio of collected negative to positive peptides was 9:1, and we maintained a similar ratio for different sequence lengths within the dataset. We compared the sequences within the negative dataset between the negative and positive datasets for an 80% sequence match and removed the ones above this threshold. Finally, we obtained a negative dataset with 792 sequences.
- Test dataset. To identify thrombin-inhibiting activity in new peptides, we collected a total of 10,743,304 peptides from the UniProt and NCBI protein databases. We searched for the source organism as one of ‘fungi’, ‘bacteria’, ‘snakes’, ‘leeches’, ‘humans’, ‘mice’, ‘eukaryota’, and ‘viruses’, and the results were filtered to a sequence length between 5 and 200 amino acids. Peptides in the sequence range of 5 to 15 amino acids were collected independently of the source. The peptides could be true peptides or random fragments of large proteins.
2.2. Feature Extraction
- Global Physico-Chemical Properties (PCP). We used the ‘Biopython ProtParam’ package to extract the global properties of the collected sequences which include sequence length, molecular weight, aromaticity, isoelectric point, and instability. This constitutes a 5-element vector.
- Amino Acid Composition (AAC). The amino acid composition (AAC) is a measure that quantifies the relative abundance of each amino acid in a peptide sequence. These features were extracted using the ‘propy3′ python package [29]. The following equation represents the amino acid composition function:
- Composition Transition Distribution (CTD). The CTD descriptor is a 147-element vector that describes different physico-chemical properties of a peptide [30] (Table S2). The physico-chemical properties covered by CTD features are ‘polarity’, ‘polarizability’, ‘charge’, ‘secondary structure’, ‘hydrophobicity’, ‘normalized van der Waals volume’, and ‘solvent accessibility’. The CTD descriptor groups the amino acids into three classes for each physico-chemical property. The composition (C) descriptor describes the global percentage of each class in a peptide sequence, the transition (T) descriptor characterizes the percent frequency of transitions between two classes in a peptide sequence, and the distribution (D) descriptor specifies the distribution patterns of each class in a sequence. These CTD properties were extracted using the ‘propy3 CTD’ package.
- Dipeptide Composition (DPC). The DPC descriptor was extracted using the ‘propy3 AAComposition’ package which returns a 400-element vector containing percent fractions of dipeptides, i.e., AA, AC, AD, …, VY, and VV, in a peptide sequence. The DPC fraction percentage is calculated as follows:
2.3. Classification Models
2.4. Clustering
2.5. Regression Models
2.6. Molecular Docking
3. Results
3.1. Characteristics of Thrombin-Inhibiting Peptides
3.2. Development of Machine Learning Models for Thrombin Inhibition
3.3. Prediction of Antithrombotic Efficacy of Peptide Hits
3.4. Prediction of Antithrombotic Activity in Test Peptides
3.5. Clustering of Hits to Identify Unique Peptides
3.6. Ranking of Top Hits Based on Binding Scores
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marcum, J.A. Defending the priority of “remarkable researches”: The discovery of fibrin ferment. Hist. Philos. Life Sci. 1998, 20, 51–76. [Google Scholar] [PubMed]
- Remiker, A.S.; Palumbo, J.S. Mechanisms coupling thrombin to metastasis and tumorigenesis. Thromb. Res. 2018, 164, S29–S33. [Google Scholar] [CrossRef]
- Aliter, K.F.; Al-Horani, R.A. Thrombin Inhibition by Argatroban: Potential Therapeutic Benefits in COVID-19. Cardiovasc. Drugs Ther. 2021, 35, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.A.; Philippou, H.; Huntington, J.A. Directing thrombin. Blood 2005, 106, 2605–2612. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.G. Thrombin formation. Chest 2003, 124 (Suppl. S3), 4S–10S. [Google Scholar] [CrossRef]
- Gustafsson, D.; Bylund, R.; Antonsson, T.; Nilsson, I.; Nyström, J.E.; Eriksson, U.; Bredberg, U.; Teger-Nilsson, A.C. A new oral anticoagulant: The 50-year challenge. Nat. Rev. Drug Discov. 2004, 3, 649–659. [Google Scholar] [CrossRef]
- Di Nisio, M.; Middeldorp, S.; Büller, H.R. Direct Thrombin Inhibitors. N. Engl. J. Med. 2005, 353, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.; Sobieraj-Teague, M.; Eikelboom, J.W. Direct oral anticoagulants: Evidence and unresolved issues. Lancet 2020, 396, 1767–1776. [Google Scholar] [CrossRef]
- Montinari, M.R.; Minelli, S. From ancient leech to direct thrombin inhibitors and beyond: New from old. Biomed. Pharmacother. 2022, 149, 112878. [Google Scholar] [CrossRef]
- Soares, T.A.; Nunes-Alves, A.; Mazzolari, A.; Ruggiu, F.; Wei, G.-W.; Merz, K. The (Re)-Evolution of Quantitative Structure–Activity Relationship (QSAR) studies propelled by the surge of machine learning methods. J. Chem. Inf. Model. 2022, 62, 5317–5320. [Google Scholar] [CrossRef] [PubMed]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Xie, X.-Q. Generative chemistry: Drug discovery with deep learning generative models. J. Mol. Model. 2021, 27, 71. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Li, A.; Zheng, H.; Yang, B.; Lu, Y. Machine learning advances in predicting peptide/protein-protein interactions based on sequence information for lead peptides discovery. Adv. Biol. 2023, 7, 2200232. [Google Scholar] [CrossRef] [PubMed]
- Syrlybaeva, R.; Strauch, E.M. Deep learning of protein sequence design of protein–protein interactions. Bioinformatics 2023, 39, btac733. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Tünnermann, L.; Löfstedt, T.; Gratz, R. Transformer-based deep learning for predicting protein properties in the life sciences. Elife 2023, 12, e82819. [Google Scholar] [CrossRef]
- Noé, F.; Tkatchenko, A.; Müller, K.-R.; Clementi, C. Machine learning for molecular simulation. Annu. Rev. Phys. Chem. 2020, 71, 361–390. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, P.; Lin, W.-Z.; Jia, J.-H.; Chou, K.-C. iAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 2013, 436, 168–177. [Google Scholar] [CrossRef]
- Manavalan, B.; Basith, S.; Shin, T.H.; Choi, S.; Kim, M.O.; Lee, G. MLACP: Machine-learning-based prediction of anticancer peptides. Oncotarget 2017, 8, 77121–77136. [Google Scholar] [CrossRef]
- Bose, B.; Downey, T.; Ramasubramanian, A.K.; Anastasiu, D.C. Identification of distinct characteristics of antibiofilm peptides and prospection of diverse sources for efficacious sequences. Front. Microbiol. 2022, 12, 783284. [Google Scholar] [CrossRef]
- Kumar, R.; Chaudhary, K.; Singh Chauhan, J.; Nagpal, G.; Kumar, R.; Sharma, M.; Raghava, G.P. An in-silico platform for predicting, screening and designing of antihypertensive peptides. Sci. Rep. 2015, 5, 12512. [Google Scholar] [CrossRef]
- Lee, E.Y.; Fulan, B.M.; Wong, G.C.L.; Ferguson, A.L. Mapping membrane activity in undiscovered peptide sequence space using machine learning. Proc. Natl. Acad. Sci. USA 2016, 113, 13588–13593. [Google Scholar] [CrossRef] [PubMed]
- Lakshmaiah Narayana, J.; Mishra, B.; Lushnikova, T.; Wu, Q.; Chhonker, Y.S.; Zhang, Y.; Zarena, D.; Salnikov, E.S.; Dang, X.; Wang, F.; et al. Two distinct amphipathic peptide antibiotics with systemic efficacy. Proc. Natl. Acad. Sci. USA 2020, 117, 19446–19454. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Sercu, T.; Wadhawan, K.; Padhi, I.; Gehrmann, S.; Cipcigan, F.; Chenthamarakshan, V.; Strobelt, H.; dos Santos, C.; Chen, P.-Y.; et al. Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations. Nat. Biomed. Eng. 2021, 5, 613–623. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Agarwala, R.; Barret, T.; Beck, J.; Benson, D.A. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Chapman, B.; Chang, J. Biopython: Python Tools for Computation Biology. 2000. Available online: http://www.bris.ac.uk/Depts/Chemistry/MOTM/ (accessed on 3 November 2023).
- Xiao, N.; Cao, D.S.; Zhu, M.F.; Xu, Q.S. Protr/ProtrWeb: R package and web server for generating various numerical representation schemes of protein sequences. In Bioinformatics; Oxford University Press: Oxford, UK, 2015; pp. 1857–1859. [Google Scholar] [CrossRef]
- Govindan, G.; Nair, A.S. Composition, Transition and Distribution (CTD)—A dynamic feature for predictions based on hierarchical structure of cellular sorting. In Proceedings of the 2011 Annual IEEE India Conference, Hyderabad, India, 16–18 December 2011; pp. 1–6. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Duchesnay, É. Scikit-learn: Machine learning in Python. J. Machine Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Randriamihamison, N.; Vialaneix, N.; Neuvial, P. Applicability and interpretability of ward’s hierarchical agglomerative clustering with or without contiguity constraints. J. Classif. 2021, 38, 363–389. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0 Contributors. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.Y. HPEPDOCK: A web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018, 46, W443–W450. [Google Scholar] [CrossRef] [PubMed]
- Kurcinski, M.; Jamroz, M.; Blaszczyk, M.; Kolinski, A.; Kmiecik, S. CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res. 2015, 43, W419–W424. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Huntington, J.A. Molecular recognition mechanisms of thrombin. J. Thromb. Haemost. 2005, 3, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Di Cera, E. Thrombin. Mol. Asp. Med. 2008, 29, 203–254. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswamy, S. Exosite-driven substrate specificity and function in coagulation. J. Thromb. Haemost. 2005, 3, 54–67. [Google Scholar] [CrossRef]
- Iyer, J.K.; Koh, C.Y.; Kazimirova, M.; Roller, L.; Jobichen, C.; Swaminathan, K.; Mizuguchi, J.; Iwanaga, S.; Nuttall, P.A.; Chan, M.Y.; et al. Avathrin: A novel thrombin inhibitor derived from a multicopy precursor in the salivary glands of the ixodid tick, Amblyomma variegatum. FASEB J. 2017, 31, 2981–2995. [Google Scholar] [CrossRef] [PubMed]
- Peeters, H. Protides of the Biological Fluids; Elsevier: Amsterdam, The Netherlands, 1975. [Google Scholar] [CrossRef]
- Ribeiro, J.M. Blood-feeding arthropods: Live syringes or invertebrate pharmacologists? Infect Agents Dis. 1995, 4, 143–152. [Google Scholar] [PubMed]
- Myles, T.; Church, F.C.; Whinna, H.C.; Monard, D.; Stone, S.R. Role of thrombin anion-binding exosite-I in the formation of thrombin-serpin complexes. J. Biol. Chem. 1998, 273, 31203–31208. [Google Scholar] [CrossRef]
- Mans, B.J.; Louw, A.I.; Neitz, A.W.H. Amino acid sequence and structure modeling of savignin, a thrombin inhibitor from the tick, Ornithodoros savignyi. Insect Biochem. Mol. Biol. 2002, 32, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Howard, N.; Abell, C.; Blakemore, W.; Chessari, G.; Congreve, M.; Howard, S.; Jhoti, H.; Murray, C.W.; Seavers, L.C.; van Montfort, R.L. Application of fragment screening and fragment linking to the discovery of novel thrombin inhibitors. J. Med. Chem. 2006, 49, 1346–1355. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.; Sali, A. Comparative protein structure modeling and its applications to drug discovery. Annu. Rep. Med. Chem. 2004, 39, 259–276. [Google Scholar] [CrossRef]
- Böhm, H.-J.; Stahl, M. Structure-based library design: Molecular modelling merges with combinatorial chemistry. Curr. Opin. Chem. Biol. 2000, 4, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Giguère, S.; Laviolette, F.; Marchand, M.; Tremblay, D.; Moineau, S.; Liang, X.; Biron, É.; Corbeil, J. Machine learning assisted design of highly active peptides for drug discovery. PLoS Comp. Biol. 2015, 11, e1004074. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Shih, N.; Yip, C.Y.C.; Li, A.W.L.; Chen, W.; Amran, F.S.; Leong, E.J.E.; Iyer, J.K.; Croft, G.; Mazlan, M.I.B.; et al. Efficacy and safety of next-genertion tick transcriptome-derived direct thrombin inhibitors. Nat. Commun. 2021, 12, 6912. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.B.; Maraganore, J.M.; Bourdon, P.; Hanson, S.R.; Harker, L.A. Antithrombotic effects of synthetic peptides targeting various functional domains of thrombin. Proc. Natl. Acad. Sci. USA 1992, 89, 6040–6044. [Google Scholar] [CrossRef]
- Hasan, A.A.; Warnock, M.; Nieman, M.; Srikanth, S.; Mahdi, F.; Krishnan, R.; Tulinsky, A.; Schmaier, A.H. Mechanisms of Arg-Pro-Pro-Gly-Phe inhibition of thrombin. Amer. J. Physiol. Heart. Circ. Physiol. 2003, 285, H183–H193. [Google Scholar] [CrossRef]
- Cheng, S.; Tu, M.; Liu, H.; An, Y.; Du, M.; Zhu, B. A novel heptapeptide derived from Crassostrea gigas shows anticoagulant activity by targeting for thrombin active domain. Food Chem. 2021, 334, 127507. [Google Scholar] [CrossRef]
- Chen, F.; Jiang, H.; Lu, Y.; Chen, W.; Huang, G. Identification and in silico analysis of antithrombotic peptides from the enzymatic hydrolysates of Tenebrio molitor larvae. Eur. Food Res. Technol. 2019, 245, 2687–2695. [Google Scholar] [CrossRef]
- Kazimtrova, M.; Kini, R.M.; Koh, C.Y. Thrombin Inhibitor. U.S. Patent 9217027, 2016. [Google Scholar]
- Liu, H.; Tu, M.; Cheng, S.; Xu, Z.; Xu, X.; Du, M. Anticoagulant decapeptide interacts with thrombin at the active site and exosite-I. J. Agric. Food Chem. 2020, 68, 176–184. [Google Scholar] [CrossRef]
- Cheng, S.; Wu, D.; Liu, H.; Xu, X.; Zhu, B.; Du, M. A novel anticoagulant peptide discovered from Crassostrea gigas by combining bioinformatics with the enzymolysis strategy: Inhibitory kinetics and mechanisms. Food Funct. 2021, 12, 10136–10146. [Google Scholar] [CrossRef] [PubMed]
- Naski, M.C.; Fenton, J.W.; Maraganore, J.M.; Olson, S.T.; Shafer, J.A. The COOH-terminal domain of hirudin. An exosite-directed competitive inhibitor of the action of alpha-thrombin on fibrinogen. J. Biol. Chem. 1990, 265, 13484–13489. [Google Scholar] [CrossRef]
- Feng, L.; Tu, M.; Qiao, M.; Fan, F.; Chen, H.; Song, W.; Du, M. Thrombin inhibitory peptides derived from Mytilus edulis proteins: Identification, molecular docking and in silico prediction of toxicity. Eur. Food Res. Technol. 2018, 244, 207–217. [Google Scholar] [CrossRef]
- Mosesson, M.W.; Meh, D.A. Thrombin Inhibitor. U.S. Patent 5985833, 2000. [Google Scholar]
- Stubbs, M.T.; Oschkinat, H.; Mayr, I.; Huber, R.; Angliker, H.; Stone, S.R.; Bode, W. The interaction of thrombin with fibrinogen. A structural basis for its specificity. Eur. J. Biochem. 1992, 206, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Scharf, M.; Engels, J.; Tripier, D. Primary structures of new iso-hirudins. FEBS Lett. 1989, 255, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Maraganore, J.M.; Bourdon, P.; Jablonski, J.; Ramachandran, K.L.; Fenton, J.W. Design and characterization of hirulogs: A novel class of bivalent peptide inhibitors of thrombin. Biochemistry 1990, 29, 7095–7101. [Google Scholar] [CrossRef]
- Ni, F.; Tolkatchev, D.; Natapova, A.; Koutychenko, A. Peptide Inhibitors of Thrombin as Potent Anticoagulants. U.S. Patent US7456152B2, 2008. [Google Scholar]
- Figueiredo, A.C.; de Sanctis, D.; Gutiérrez-Gallego, R.; Cereija, T.B.; Macedo-Ribeiro, S.; Fuentes-Prior, P.; Pereira, P.J. Unique thrombin inhibition mechanism by anophelin, an anticoagulant from the malaria vector. Proc. Natl. Acad. Sci. USA 2012, 109, E3649–E3658. [Google Scholar] [CrossRef]
- Cappello, M.; Li, S.; Chen, X.; Li, C.B.; Harrison, L.; Narashimhan, S.; Beard, C.B.; Aksoy, S. Tsetse thrombin inhibitor: Bloodmeal-induced expression of an anticoagulant in salivary glands and gut tissue of Glossina morsitans morsitans. Proc. Natl. Acad. Sci. USA 1998, 95, 14290–14295. [Google Scholar] [CrossRef]
- Koh, C.Y.; Kazimirova, M.; Trimnell, A.; Takac, P.; Labuda, M.; Nuttall, P.A.; Kini, R.M. Variegin, a novel fast and tight binding thrombin inhibitor from the tropical bont tick. J. Biol. Chem. 2007, 282, 29101–29113. [Google Scholar] [CrossRef]
- Giri, P.K.; Tang, X.; Thangamani, S.; Shenoy, R.T.; Ding, J.L.; Swaminathan, K.; Sivaraman, J. Modifying the substrate specificity of Carcinoscorpius rotundicauda serine protease inhibitor domain 1 to target thrombin. PLoS ONE 2010, 5, e15258. [Google Scholar] [CrossRef]
- Sarmientos, P.; Poet, P.D.T.D.; Nitti, G.; Scacheri, E. Antithrombin Polypeptides. U.S. Patent US5439820A, 1995. [Google Scholar]
- Hong, S.J.; Kang, K.W. Purification of granulin-like polypeptide from the blood-sucking leech, Hirudo nipponia. Protein Expr. Purif. 1999, 16, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Y.; Lu, Z.; Zhai, L.; Jiang, J.; Liu, J.; Yu, H. A novel serine protease inhibitor from the venom of Vespa bicolor Fabricius. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2009, 153, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Schlott, B.; Wöhnert, J.; Icke, C.; Hartmann, M.; Ramachandran, R.; Gührs, K.H.; Glusa, E.; Flemming, J.; Görlach, M.; Grosse, F.; et al. Interaction of Kazal-type inhibitor domains with serine proteinases: Biochemical and structural studies. J. Mol. Biol. 2002, 318, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Strube, K.H.; Kröger, B.; Bialojan, S.; Otte, M.; Dodt, J. Isolation, sequence analysis, and cloning of haemadin. An anticoagulant peptide from the Indian leech. J. Biol. Chem. 1993, 268, 8590–8595. [Google Scholar] [CrossRef] [PubMed]
- Brahma, R.K.; Blanchet, G.; Kaur, S.; Kini, R.M.; Doley, R. Expression and characterization of haemathrins, madanin-like thrombin inhibitors, isolated from the salivary gland of tick Haemaphysalis bispinosa (Acari: Ixodidae). Thromb. Res. 2017, 152, 20–29. [Google Scholar] [CrossRef]
- Clayton, D.; Kulkarni, S.S.; Sayers, J.; Dowman, L.J.; Ripoll-Rozada, J.; Pereira, P.J.; Payne, R.J. Chemical synthesis of a haemathrin sulfoprotein library reveals enhanced thrombin inhibition following tyrosine sulfation. RSC Chem. Biol. 2020, 1, 379–384. [Google Scholar] [CrossRef]
- Jablonka, W.; Kotsyfakis, M.; Mizurini, D.M.; Monteiro, R.Q.; Lukszo, J.; Drake, S.K.; Ribeiro, J.M.; Andersen, J.F. Identification and mechanistic analysis of a novel tick-derived inhibitor of thrombin. PLoS ONE 2015, 10, e0133991. [Google Scholar] [CrossRef]
- Thompson, R.E.; Liu, X.; Ripoll-Rozada, J.; Alonso-García, N.; Parker, B.L.; Pereira, P.J.B.; Payne, R.J. Tyrosine sulfation modulates activity of tick-derived thrombin inhibitors. Nature Chem. 2017, 9, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, S.; Okada, M.; Isawa, H.; Morita, A.; Yuda, M.; Chinzei, Y. Identification and characterization of novel salivary thrombin inhibitors from the ixodidae tick, Haemaphysalis longicornis. Eur. J. Biochem. 2003, 270, 1926–1934. [Google Scholar] [CrossRef] [PubMed]
- Krstenansky, J.L.; Owen, T.J.; Yates, M.T.; Mao, S.J.T. The C-terminal binding domain of hirullin P18. FEBS Lett. 1990, 269, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Steiner, V.; Knecht, R.; Börnsen, K.O.; Gassmann, E.; Stone, S.R.; Raschdorf, F.; Schlaeppi, J.M.; Maschler, R. Primary structure and function of novel O-glycosylated hirudins from the leech Hirudinaria manillensis. Biochemistry 1992, 31, 2294–2298. [Google Scholar] [CrossRef] [PubMed]
- Scacheri, E.; Nitti, G.; Valsasina, B.; Orsini, G.; Visco, C.; Ferrera, M.; Sawyer, R.T.; Sarmientos, P. Novel hirudin variants from the leech Hirudinaria manillensis. Amino acid sequence, cDNA cloning and genomic organization. Eur. J. Biochem. 1993, 214, 295–304. [Google Scholar] [CrossRef]
- Rydel, T.J.; Ravichandran, K.G.; Tulinsky, A.; Bode, W.; Huber, R.; Roitsch, C.; Fenton, J.W., 2nd. The structure of a complex of recombinant hirudin and human alpha-thrombin. Science 1990, 249, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.R.; Hofsteenge, J. Kinetics of the inhibition of thrombin by hirudin. Biochemistry 1986, 25, 4622–4628. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E. Bivalent direct thrombin inhibitors: Hirudin and bivalirudin. Best Pract. Res. Clin. Haematol. 2004, 17, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.M.O.; Tanaka-Azevedo, A.M.; Araujo, M.S.; Juliano, M.A.; Tanaka, A.S. Characterization of thrombin inhibitory mechanism of rAaTI, a Kazal-type inhibitor from Aedes aegypti with anticoagulant activity. Biochimie 2011, 93, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Salzet, M.; Chopin, V.; Baert, J.; Matias, I.; Malecha, J. Theromin, a novel leech thrombin inhibitor. J. Biol. Chem. 2000, 275, 30774–30780. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Liu, F.; Guo, Q.; Lu, Y.; Shi, H.; Ding, A.; Xu, C. Identification and characterization of hirudin-HN, a new thrombin inhibitor, from the salivary glands of Hirudo nipponia. PeerJ 2019, 7, e7716. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, C.; Imamura, S.; Konnai, S.; Yamada, S.; Nishikado, H.; Ohashi, K.; Onuma, M. A novel gene encoding a thrombin inhibitory protein in a cDNA library from Haemaphysalis longicornis salivary gland. J. Vet. Med. Sci. 2006, 68, 447–452. [Google Scholar] [CrossRef][Green Version]
- Zhang, D.; Cupp, M.S.; Cupp, E.W. Thrombostasin: Purification, molecular cloning and expression of a novel anti-thrombin protein from horn fly saliva. Insect Biochem. Mol. Biol. 2002, 32, 321–330. [Google Scholar] [CrossRef]
- Pirone, L.; Ripoll-Rozada, J.; Leone, M.; Ronca, R.; Lombardo, F.; Fiorentino, G.; Andersen, J.F.; Pereira, P.J.; Arcà, B.; Pedone, E. Functional analyses yield detailed insight into the mechanism of thrombin inhibition by the antihemostatic salivary protein CE5 from Anopheles gambiae. J. Biol. Chem. 2017, 292, 12632–12642. [Google Scholar] [CrossRef] [PubMed]
- Campos, I.T.; Amino, R.; Sampaio, C.A.; Auerswald, E.A.; Friedrich, T.; Lemaire, H.G.; Schenkman, S.; Tanaka, A.S. Infestin, a thrombin inhibitor presents in Triatoma infestans midgut, a Chagas’ disease vector: Gene cloning, expression and characterization of the inhibitor. Insect Biochem. Mol. Biol. 2002, 32, 991–997. [Google Scholar] [CrossRef]
- Friedrich, T.; Kröger, B.; Bialojan, S.; Lemaire, H.G.; Höffken, H.W.; Reuschenbach, P.; Otte, M.; Dodt, J. A Kazal-type inhibitor with thrombin specificity from Rhodnius prolixus. J. Biol. Chem. 1993, 268, 16216–16222. [Google Scholar] [CrossRef] [PubMed]
- Mende, K.; Petoukhova, O.; Koulitchkova, V.; Schaub, G.A.; Lange, U.; Kaufmann, R.; Nowak, G. Dipetalogastin, a potent thrombin inhibitor from the blood-sucking insect Dipetalogaster maximus cDNA cloning, expression and characterization. Eur. J. Biochem. 1999, 266, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Nienaber, J.; Gaspar, A.R.M.; Neitz, A.W.H. Savignin, a potent thrombin inhibitor isolated from the salivary glands of the tick Ornithodoros savignyi (Acari: Argasidae). Exp. Parasitol. 1999, 93, 82–91. [Google Scholar] [CrossRef]
- van de Locht, A.; Stubbs, M.T.; Bode, W.; Friedrich, T.; Bollschweiler, C.; Höffken, W.; Huber, R. The ornithodorin-thrombin crystal structure, a key to the TAP enigma? EMBO J. 1996, 15, 6011–6017. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Zhou, J.; Gong, H.; Boldbaatar, D.; Shirafuji, R.; Battur, B.; Nishikawa, Y.; Fujisaki, K. Hemalin, a thrombin inhibitor isolated from a midgut cDNA library from the hard tick Haemaphysalis longicornis. J. Insect Physiol. 2009, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Carvalho, A.L.; Guimarães, P.R.; Abreu, P.A.; Dutra, D.L.S.; Junqueira-de-Azevedo, I.L.M.; Rodrigues, C.R.; Ho, P.L.; Castro, H.C.; Zingali, R.B. Identification and characterization of a new member of snake venom thrombin inhibitors from Bothrops insularis using a proteomic approach. Toxicon 2008, 51, 659–671. [Google Scholar] [CrossRef]
- Macedo-Ribeiro, S.; Almeida, C.; Calisto, B.M.; Friedrich, T.; Mentele, R.; Stürzebecher, J.; Fuentes-Prior, P.; Barbosa Pereira, P.J. Isolation, cloning and structural characterization of Boophilin, a multifunctional kunitz-type proteinase inhibitor from the cattle tick. PLoS ONE 2008, 3, e1624. [Google Scholar] [CrossRef]
- Mans, B.J.; Andersen, J.F.; Schwan, T.G.; Ribeiro, J.M.C. Characterization of anti-hemostatic factors in the argasid, Argas monolakensis: Implications for the evolution of blood-feeding in the soft tick family. Insect Biochem. Mol. Biol. 2008, 38, 22–41. [Google Scholar] [CrossRef]
- Noeske-Jungblut, C.; Haendler, B.; Donner, P.; Alagon, A.; Possani, L.; Schleuning, W.-D. Triabin, a highly potent exosite inhibitor of thrombin. J. Biol. Chem. 1995, 270, 28629–28634. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Takeuchi, H.; Jonczy, J.; Rees, H.H.; Turner, P.C. A thrombin inhibitor from the ixodid tick, Amblyomma hebraeum. Gene 2004, 342, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Hengst, U.; Albrecht, H.; Hess, D.; Monard, D. The Phosphatidylethanolamine-binding protein is the prototype of a novel family of serine protease inhibitors. J. Biol. Chem. 2001, 276, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Eigenbrot, C.; Liang, W.-C.; Stawicki, S.; Shia, S.; Fan, B.; Ganesan, R.; Lipari, M.T.; Kirchhofer, D. Structural insight into distinct mechanisms of protease inhibition by antibodies. Proc. Natl. Acad. Sci. USA 2007, 104, 19784–19789. [Google Scholar] [CrossRef] [PubMed]
- Arocas, V.; Castro, H.C.; Zingali, R.B.; Guillin, M.C.; Jandrot-Perrus, M.; Bon, C.; Wisner, A. Molecular cloning and expression of bothrojaracin, a potent thrombin inhibitor from snake venom. Eur. J. Biochem. 1997, 248, 550–557. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Feature Reduction Method | Number of Features |
|---|---|---|
| SVC Linear | RFE | 120 |
| SVC RBF | SFS | 314 |
| Logistic Regression | RFE | 54 |
| Random Forest | RFE | 508 |
| KNN | SFS | 257 |
| XGBoost | RFE | 32 |
| Model | Stage | Log Training RMSE | Log Validation RMSE | Log Test RMSE |
|---|---|---|---|---|
| SVR with Linear Kernel | Baseline | 0.715 | 1.951 | 1.47 |
| SFS with 51 features | 0.778 | 1.149 | 1.221 | |
| SVR with RBF Kernel | Baseline | 0.279 | 1.847 | 1.541 |
| SFS with 125 features | 0.2 | 1.114 | 1.06 | |
| Lasso Regression | Baseline | 1.14 | 1.887 | 1.802 |
| SFS with 28 features | 1.388 | 1.728 | 1.107 |
| Peptide | Sequence | Source | KD (nM) | Docking Scores | Binding Residues | Binding Sites |
|---|---|---|---|---|---|---|
| T49 | QGNRKTTKEGSNDL | Homo sapiens (cytokine-dependent hematopoietic cell linker isoform X1) | 9 | −175.365 | S20, D21, A22, E23, I24, G25, M26, P28, K70, H71, E80, D116, Y117, I118, Y134, K135, R137, V158, N159, E185, K202, S203, P204, N205, R206, W207 | Exosite 1 |
| T34 | EYEEVEASPEKET | Meleagris gallopavo (tubulin beta-1 chain) | 12 | −166.309 | I47, S48, W51, K87, Y89, I90, H91, P92, R93, L105, K107, K109, K110, P111, V112, C122, L123, R126, E127, F232, K236, K240 | Exosite 2 |
| T45 | SGEGSFQPSQQNPQ | Triticum aestivum (gliadin peptide) | 16 | −180.56 | F34, R35, K36, S37, P38, Q38A, E40, R67, K70, H71, R73, T74, R75, Y76, E77, R77a, N78, I79, W141, N143, L144, Q151, P152, S153 | Active site and Exosite 1 |
| T56 | ARATAETDATANRG | Mycobacterium tuberculosis (prophage protein) | 20 | −175.107 | E23, I24, K36, P38, K70, H71, S72, R73, T74, R75, E77, E80, S153, V154 | Exosite 1 |
| T52 | EPTTEDLYFQSDND | M13 helper phage (pIII) | 31 | −189.841 | N98, D100, R101, T147, R173, R175, T177, E217, R232 | Active site and Exosite 2 |
| T33 | IYRFEPSKFIGE | Nymphaea colorata (unnamed protein) | 37 | −213.129 | E39, L40, R93, E98, N143, S171, R173, I174, R175, I176, E192, E217, A221D | Active site |
| T39 | ACENEDFEGIPGEA | Homo sapiens (hirugen, synthetic construct) | 150 | −168.785 | S20, D21, A22, E23, I24, G25, M26, P28, W29, I68, G69, K70, I79, E80, K81, A113, F114, S115, D116, K135, G149a, K149b, V157, V158, N159, E184a, G186c, K186c, K202, S203, P204, R204a | Exosite 1 |
| T57 | FEFEFEPGGGRGDS | Spirochaetales bacterium (SpoIIE family protein phosphatase) | 170 | −209.705 | K36, S37, P38, Q39, E40, W60a, S72, R73, T74, R75, Y76, R97, E98, N99, N143, L144, K145, W147, T148, Q151, S153, C191, D221a | Active site and Exosite 1 |
| T54 | RYEVRAELPGVDPD | Mycobacterium tuberculosis (erythromycin esterase) | 240 | −203.537 | E23, M32, R35, K70, H71, R73, T74, R75, Y76, V154, Q156 | Exosite 1 |
| T27 | VQIYEEARKFS | Potamochoerus porcus (DEAD-box protein 3) | 480 | −187.722 | H91, P92, R93, Y94, L99, D100, R101, D125, I176, T177, N179, H230, V231, F232, R233, L234, W237, I238, I242, D243 | Exosite 2 |
| T44 | GNTRTAESGDEDFF | Eubacteriales bacterium (transglycosylase domain-containing protein) | 530 | −181.246 | R35, P38, Q39, E40, R67, K70, H71, S72, R73, T74, Y76, E77, R78, N79, E80, G142, N143, L144, Q151, P152, S153, V154, E192 | Active site and Exosite 1 |
| T55 | NRLVQNPPKKFSGE | Burkholderia sp. Bp9140 (hypothetical protein) | 610 | −224.973 | S20, D21, A22, E23, Q39, V67, H71, S72, T74, Y76, S116, Y117, K135, W141, A149V, S153, V154, L155, V157, N158, E185, R187, K202, S203, P204, R206 | Exosite 1 |
| T31 | AEYETVQNSFNQ | Cellvibrio fibrivorans (cellulase family glycosyl hydrolase) | 630 | −185.041 | P92, R93, W96, N98, L99, D100, R101, D102, I103, R126, A129A, B129S, Q131, E164, R175, I176, T177, D178, N179, H230, F232, R233, K236, Q244 | Exosite 2 |
| T46 | SSGSVGESSSKGPR | Pan pansicus (cytokeratin-10) | 630 | −182.793 | E23, I24, G25, F34, R35, S37, P38, Q39, E40, L41, D60W, K70, H71, S72, R73, N79, E98, N99, D116, N143, L144, K145, P152, S153, L155, Q156, E192, W215, G216, E217 | Active site and Exosite 1 |
| T40 | VQGSDQSDSANVQR | Hoeflea sp. (UDP N-acetylmuramate L-alanine ligase) | 770 | −175.557 | I23, F34, R35, K36, S37, P38, Q39, E40, L42, R73, W140, N143, L144, E146, C149V, P152, S153, E192, E216, G218, C219, D220, R221 | Active site |
| T41 | NDDEDPKSHRDPSN | FGF-4 synthetic construct | 1200 | −209.886 | I24, G25, Q30, K70, R78, N79, I80, E80, K81, I82, K107, L108, K109, K110, P111, V112, F114, Y117, I118, H119 | Exosite 1 |
| T32 | GEKPDEFESGSP | Poecilia Mexicana (ribosomal protein S7) | 1300 | −194.088 | R101, R126, T128, A129A, S130, L132, Q133, E164, R165, K169, D178, N179, M180, S203, P204, F205, H230, R233, K236 | Exosite 2 |
| T42 | RGNNDIGSGFNDDP | Cellulomonas soli (glycosyl transferase) | 1600 | −185.149 | N95, W96, E97, N98, L99, D100, V163, P166, K169, D170, S171, T172, I174, R175, I176, Y184a, K184b, E185, E217, R221b, D222, G223, K224 | Exosite 2 |
| T48 | GIGPKFQHSGGEPP | Mycobacterium tuberculosis (prophage protein) | 1800 | −205.784 | I90, H91, R93, Y94, N95, N99, L100, D100, R173, I174, R175, I176, F227, V241, I242, F245, E246 | Exosite 2 |
| T29 | MEEGPSDPGSRS | Mogibacterium sp. (haloacid dehalogenase-like hydrolase) | 1800 | −170.831 | I24, G25, Q30, K70, R78, N79, I80, E80, K81, I82, K107, L108, K109, K110, P111, V112, F114, Y117, I118, H119 | Exosite 2 |
| T43 | HGEGTFTSDLSKQM | Heloderma suspectum (exendin 4 venom) | 2500 | −190.877 | E23, I24, G25, M26, E39, K70, H71, E77, R77a, N78, I79, D116, I118, H119, N143, S153, V154, Q156 | Exosite 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balakrishnan, N.; Katkar, R.; Pham, P.V.; Downey, T.; Kashyap, P.; Anastasiu, D.C.; Ramasubramanian, A.K. Prospection of Peptide Inhibitors of Thrombin from Diverse Origins Using a Machine Learning Pipeline. Bioengineering 2023, 10, 1300. https://doi.org/10.3390/bioengineering10111300
Balakrishnan N, Katkar R, Pham PV, Downey T, Kashyap P, Anastasiu DC, Ramasubramanian AK. Prospection of Peptide Inhibitors of Thrombin from Diverse Origins Using a Machine Learning Pipeline. Bioengineering. 2023; 10(11):1300. https://doi.org/10.3390/bioengineering10111300
Chicago/Turabian StyleBalakrishnan, Nivedha, Rahul Katkar, Peter V. Pham, Taylor Downey, Prarthna Kashyap, David C. Anastasiu, and Anand K. Ramasubramanian. 2023. "Prospection of Peptide Inhibitors of Thrombin from Diverse Origins Using a Machine Learning Pipeline" Bioengineering 10, no. 11: 1300. https://doi.org/10.3390/bioengineering10111300
APA StyleBalakrishnan, N., Katkar, R., Pham, P. V., Downey, T., Kashyap, P., Anastasiu, D. C., & Ramasubramanian, A. K. (2023). Prospection of Peptide Inhibitors of Thrombin from Diverse Origins Using a Machine Learning Pipeline. Bioengineering, 10(11), 1300. https://doi.org/10.3390/bioengineering10111300