Abstract
Despite extensive genetic and biochemical characterization, the molecular genetic basis underlying the biosynthesis of β-diketones remains largely unexplored. β-Diketones and their complexes find broad applications as biologically active compounds. In this study, in silico molecular docking results revealed that two β-diketone derivatives, namely 2-(2-(4-fluorophenyl)hydrazono)-5,5-dimethylcyclohexane-1,3-dione and 5,5-dimethyl-2-(2-(2-(trifluoromethyl)phenyl)hydrazono)cyclohexane-1,3-dione, exhibit anti-COX-2 activities. However, recent docking results indicated that the relative anti-COX-2 activity of these two studied β-diketones was influenced by the employed docking programs. For improved design of COX-2 inhibitors from β-diketones, we conducted molecular dynamics simulations, density functional theory (DFT) calculations, Hirshfeld surface analysis, energy framework, and ADMET studies. The goal was to understand the interaction mechanisms and evaluate the inhibitory characteristics. The results indicate that 5,5-dimethyl-2-(2-(2-(trifluoromethyl)phenyl)hydrazono)cyclohexane-1,3-dione shows greater anti-COX-2 activity compared to 2-(2-(4-fluorophenyl)hydrazono)-5,5-dimethylcyclohexane-1,3-dione.
1. Introduction
Currently, the realm of β-diketones stands as a captivating focus of exploration owing to its noteworthy biological impact [1,2,3]. Analytical chemistry frequently employs β-diketones as a group of spectrophotometric reagents due to their remarkable ability to form complexes [4,5,6,7,8,9]. Additionally, derivatives of these compounds play a pivotal role in the treatment of inflammatory diseases with antioxidant and antiviral attributes [10,11,12]. Various β-diketones and their complexes serve as biologically active compounds [10,12]. In addition to substituted reagents and laser chelates [13], the chemical and photochemical catalysts [14] derived from these compounds are valuable in managing inflammatory diseases.
Cyclooxygenase-2, COX-2, facilitates the conversion of arachidonic acid into prostaglandin (PGH2). PGH2, a crucial precursor of prostacyclin, is expressed during inflammation. While COX-2 remains unexpressed under normal conditions in most cells, elevated levels are observed in cells where prostaglandins are upregulated during inflammation. Moreover, COX-2 expression is heightened in many cancers, possibly due to the conversion of its product, PGH2, into prostaglandin E2 by prostaglandin E2 synthase. This compound can stimulate cancer progression. Consequently, inhibiting COX-2 could offer benefits in preventing and treating cancer [15,16].
The effectiveness of nonsteroidal anti-inflammatory drugs (NSAIDs) lies in their inhibition of prostaglandin production by COX-1 and COX-2. NSAIDs that selectively inhibit COX-2 are less likely to induce gastrointestinal side effects compared to conventional drugs. However, they may contribute to cardiovascular events such as heart failure, myocardial infarction, and stroke. Numerous pieces of evidence suggest that this is linked to the suppression of COX-2-dependent cardioprotective prostaglandins, particularly prostacyclin [17]. The selective COX-2 inhibitors can selectively inhibit the COX-2 enzyme without inhibiting COX-1 due to the structural difference of COX-1 with respect to COX-2. For example, COX-1 contains isoleucine (Ile523) in its critical position, whereas COX-2 features valine (Val523) at the same site. Although such structural difference might be minor, it could result in substantial consequences, such as the binding packet of COX-2 being larger than that of COX-1, a secondary binding packet being unlocked, and the polar amino acid ARG513 being uncovered [18].
Moreover, except for aspirin (acetylsalicylic acid), which can irreversibly bind to the serine in the active center of cyclooxygenase through its acetyl group, thereby irreversibly inhibiting cyclooxygenase activity, the inhibitory effect of most NSAIDs on cyclooxygenase is reversible and therefore weak. Since this is the case, research on COX-2 inhibitors to replace NSAIDs has drawn attention [19]. This study presented the syntheses, characterizations, DFT studies, Hirshfeld surfaces, and energy frameworks of two new β-diketone derivatives. Moreover, an in silico investigation was performed into their capacities to behave as COX-2 inhibitors.
2. Materials and Methods
2.1. Chemistry
With this section, Scheme 1, Reaction 1 demonstrates the synthesis of target compound (1) [2-(2-(4-fluorophenyl)hydrazono)-5,5-dimethylcyclohexane-1,3-dione] by employing the diazotization of different aromatic amine by using 5,5-dimethylcyclohexane-1,3-dione. As a further output of the research, 5,5-dimethyl-2-(2-(2-(trifluoromethyl)phenyl)hydrazono)cyclohexane-1,3-dione (2) had been synthesized (Scheme 1, Reaction 2).
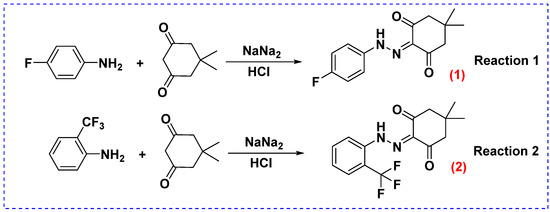
Scheme 1.
The synthesis of 2-(2-(4-fluorophenyl)hydrazono)-5,5-dimethylcyclohexane-1,3-dione (1) and 5,5-dimethyl-2-(2-(2-(trifluoromethyl)phenyl)hydrazono)cyclohexane-1,3-dione (2).
2.2. General
The response and purity of the substances were tracked using TLC (Sorbil). The examination of the substance structures was conducted employing the “Bruker APEX II CCD” diffractometer, operating at a temperature of 100 K, with λMoKα-radiation, a graphite monochromator, and both ϕ- and ω-scanning capabilities, reaching a maximum 2θ value of 560. NMR spectra were recorded on Jeol 400 Mhz NMR spectrometer.
General Procedure for the Synthesis of 2-(2-(Aryl)hydrazono)-5,5-dimethylcyclohexane-1,3-diones (1 and 2)
- 2-(2-(4-fluorophenyl)hydrazono)-5,5-dimethylcyclohexane-1,3-dione (1)
In a three-necked flask, 6.25 mmol (0.69 g) of an aromatic amine and 6.25 mmol (0.35 g) of potassium hydroxide (KOH) were dissolved in distilled water. Subsequently, 0.0225 mol (1.55 g) of sodium nitrite (NaNO2) was dissolved in 2 mL of distilled water and introduced into the mixture, which was then subjected to stirring under the influence of a magnetic stirrer. To this mixture, 2 mL of hydrochloric acid (HCl) was added dropwise, maintaining a temperature of 0 °C, and the stirring continued for 30 min.
Following this, 6.25 mmol (0.88 g) of 5,5-dimethylcyclohexane-1,3-dione and 6.25 mmol (0.51 g) of sodium acetate (CH3COONa) were dissolved in 10 mL of ethyl alcohol (C2H5OH). The temperature of this new mixture was lowered to 0 °C, and it was added drop by drop to the previously prepared mixture, which was left to stir for an additional hour at 0 °C. The resulting product was then filtered and subjected to recrystallization in ethanol. (Yield: 74%), Tm.p = 198–200 °C, C14H15N2O2F; calculated for (%): C 64.12; H 5.72; N 10.68; F 7.25. Found (%): C 64.21; H 5.59; N 10.71, F 7.15. 1H-NMR (400 MHz) (DMSO-d6) δ: ppm; 0.97–1.09 (6H, 2CH3), 2.64–2.71 (4H, 2CH2), 6.61–6.99 (4H, CH-Ph), 8.21 (1H, NH). 13C-NMR (DMSO-d6) δ: m.h.; 26.92 (2CH3), 30.61 (C), 50.51, 51.12 (2CH2), 119.9, 116.58 (4CH, Ph), 137.91 (C=N), 137.9 (C-NH), 156.92 (C-F), 187.23 (2CO).
- 5,5-dimethyl-2-(2-(2-(trifluoromethyl)phenyl)hydrazono)cyclohexane-1,3-dione (2)
In a three-necked flask, 6.25 mmol (1.00 g) of an aromatic amine and 6.25 mmol (0.35 g) of potassium hydroxide (KOH) were dissolved in distilled water. Concurrently, 0.0225 mol (1.55 g) of sodium nitrite (NaNO2) was dissolved in 2 mL of distilled water and subsequently introduced into the mixture, followed by stirring under the influence of a magnetic stirrer. A solution of 2 mL of hydrochloric acid (HCl) was added dropwise to the mixture, and the stirring continued for a duration of 30 min under a temperature of 0 °C.
Subsequently, a solution comprising 6.25 mmol (0.88 g) of 5,5-dimethylcyclohexane-1,3-dione and 6.25 mmol (0.51 g) of sodium acetate (CH3COONa) in 10 mL of ethyl alcohol (C2H5OH) was prepared. The temperature of this solution was reduced to 0 °C, and it was gradually added drop by drop to the previously established mixture, which was left to stir for an additional hour under a temperature of 0 °C. The resultant product was subjected to filtration and subsequently underwent recrystallization in ethanol. (Yield: 73%), Tm.p. = 100–102 °C. C15H15N2O2F3; calculated for (%): C 57.69; H 4.80; N 8.97; F 18.26. Found (%): C 57.79; H 4.65; N 8.73; F 18.42. 1H-NMR (400 MHz) (DMSO-d6) δ: ppm; 0.97–1.06 (6H, 2CH3), 2.64–2.69 (4H, 2CH2), 6.55–7.49 (4H, CH-Ph), 8.16 (1H, NH). 13C-NMR (DMSO-d6) δ: m.h.; 26.71, 27.12 (2CH3), 29.94 (2C), 50.2, 51.6 (2CH2), 119.1, 133.2 (4CH, Ph), 135.49 (C-CF3), 137.34 (C-NH), 137.72 (C=N), 125.4 (CF3), 187.01 (2CO).
2.3. The DFT Optimizations
The gas-phase structure of the title compounds was optimized using the density functional theory. The DFT was calculated utilizing the B3LYP hybrid functional that is based on Becke’s idea of mixing the DFT exchange with the exact one (HF) using the B3 functional and combining the LYP correlation functional [20,21]. The B3LYP calculation was performed with the 6-311++G** basis set [22]. A similar theoretical level has been proved to obtain reliable results about the thiophene carboxamide derivatives [23]. Following the acquisition of the converged geometry, the harmonic vibrational frequencies were computed at the same computational level to verify that the imaginary frequency associated with the stationary point is zero. Both the optimization of the gas-phase geometry and the analysis of the vibrational frequencies were performed using the Gaussian 16 suite program [24].
2.4. The Hirshfeld Surface and Energy Framework Analysis
The delineation of a molecule within the condensed phase and the identification of distinct entities in molecular liquids and crystals represent fundamental concepts in chemistry. Leveraging Hirshfeld’s partitioning scheme, Spackman introduced a method for partitioning the electron distribution around a crystal into molecular fragments [25,26,27,28]. The proposed method could determine intermolecular interactions between specific molecules or within crystal structures. Since it has been raised from the Hirshfeld partition, the resulting surface is thus called the Hirshfeld surface. Lately, the energy framework has emerged as a potent tool for enhancing the clarity of understanding regarding how molecules arrange themselves in a crystal. In this review, both Hirshfeld surface analysis and the energy framework of the title compound were executed utilizing the CrystalExplorer program [29].
2.5. Molecular Docking
The structural and chemical features of the two studied β-diketones, which may inhibit COX-2, were discovered using theoretical computation in this contribution. The COX-2 enzyme structure was downloaded in the PDB format from the Protein Data Bank website (PDB code 4PH9). Since several references have already shown that the used molecular docking software might affect the accuracy of the result [30,31,32,33], three molecular docking tools, iGemDock, Autodock 4, Glide and Autodock vina, were then used [34,35,36,37,38]. The Autodock 4 and Autodock vina dockings were conducted with the aid of the AMDock program [39].
2.5.1. The Preparations of the Ligands
The DFT-calculated results of the respective ligand was converted into the .mol file by the gOpenBabel [40].
2.5.2. The Preparations of the Targets
The .pdb format of the target was downloaded from the Protein Data Bank website and then treated by the UCSF Chimera 1.16 to delete the co-crystal ligands and water molecules [41,42]. Notably, one of the co-crustal ligands in the chosen COX-2 structure was ibuprofen (IBF) (see Figure 1). IBF is a NSAID commonly used to relieve pain, reduce fever, and reduce inflammation, and it was cited as the reference to investigate the inhibitory efficiencies of the studied β-diketones toward COX-2.
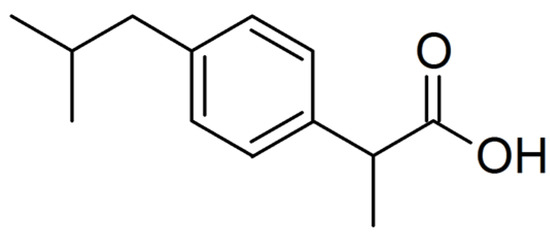
Figure 1.
The structure of ibuprofen (IBF).
2.5.3. Protocol Used for Docking Using Glide
We conducted molecular docking studies using Schrödinger Maestro Molecular Modeling software (Glide, Schrodinger, LLC, New York, NY, USA, 2023). The crystal structure of PREP (PDB: 4PH9) was chosen for its high resolution (1.81 Å) and the presence of co-crystallized ligand IBF, aiding binding site identification. Protein preparation involved the Maestro Protein Preparation Wizard (Maestro 13.2, Schrodinger, LLC, NY, 2023) with default preprocessing settings. Exceptions included missing loops and sidechains built using Prime, and “Epik” generated heteroatom states at pH 7.4. We eliminated acetate ions and glycerol, assigned hydrogen bonds, removed waters with <3 hydrogen bonds, and minimized the structure with the OPLS3e force field. Ligands, prepared with LigPrep using the OPLS-2005 force field, underwent no tautomer or stereoisomer generation. Docking utilized Glide with XP-peptide precision, employing receptor grids tailored for peptide docking with dimensions set at 15 Å (X and Y) and 10 Å (Z) around the ligand center. Ligands remained flexible, and the OPLS-2005 force field was applied throughout the process.
2.6. The ADMET Study
ADME is an acronym for “Absorption, Distribution, Metabolism, and Elimination” in pharmacokinetics and pharmaceutics, which describes the distribution of drugs in an organism. ADME affects the levels of drugs and the exposure of drugs in the tissues, which affects the potency and bioactivity of compounds in drugs. In this study, the AMDE study was conducted in SwissADME [43].
2.7. Molecular Dynamics Simulation
In our molecular dynamics simulation (MDS) analysis, simulations were executed for the COX-2:1 and COX-2:2 complexes employing the “Desmond” module (Desmond, Schrödinger, Inc., New York, NY, USA, 2023, Free academic version) for a duration of 150 nanoseconds. The simulations utilized an explicit solvent model, incorporating TIP3P water molecules, and employed the OPLS-2005 force field. The simulation was performed within a periodic boundary solvation box with dimensions of 10 Å × 10 Å × 10 Å. To maintain a charge balance of 0.15 M, sodium ions (Na+) and NaCl solutions were introduced to mimic physiological conditions. This comprehensive approach followed the MD protocol from the previous studies [44,45,46,47,48,49].
3. Results
3.1. The Crystal Structures of the Studied Compounds
The triclinic structure of 2-(2-(4-fluorophenyl)hydrazono)-5,5-dimethylcyclohexane-1,3-dione (1) was deposited at the Cambridge Crystallographic Data Centre (CCDC 1475293) and depicted as shown in Figure 2. The lattice parameters of crystal a = 5.993(2) Å, b = 10.446(4) Å, c = 10.731(4) Å, α = 97.765(8)°, β = 102.860(8)°, γ = 98.925(8)°, space group P-1, Z = 2; V = 637.0(4) Å3, Dx = 1.368 g/cm3, μ = 0.102 mm−1. Crystal sizes 0.330 × 0.260 × 0.220 mm. The molecular structure of compound (1) is shown below.
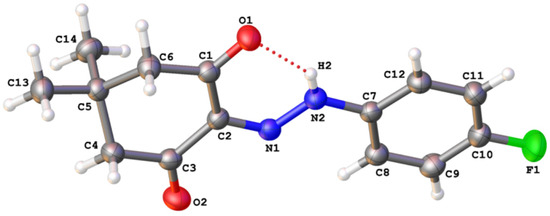
Figure 2.
Molecular structure of 2-(2-(4-fluorophenyl)hydrazono)-5,5-dimethylcyclohexane-1,3-dione (1).
The monoclinic structure of 5,5-dimethyl-2-(2-(2-(trifluoromethyl)phenyl)hydrazono)cyclohexane-1,3-dione (2) was deposited at the Cambridge Crystallographic Data Centre (CCDC 1484656) and depicted as shown in Figure 3. The lattice parameters of crystal a = 15.5610(12) Å, b = 6.1069(5) Å, c = 15.6267(12) Å, β = 97.3588(13)°, V = 1472.8(2) Å3, Z = 4, space group P21/n, Dx = 1.315 g/cm3, μ = 0.120 mm−1. Crystal sizes 0.630 × 0.220 × 0.150 mm.
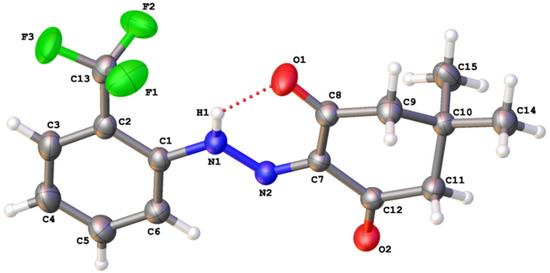
Figure 3.
Molecular structure of 5,5-dimethyl-2-(2-(2-(trifluoromethyl)phenyl)hydrazono)cyclohexane-1,3-dione (2).
3.2. The DFT-B3LYP Study
In order to elucidate the connection between intrinsic electronic properties and the chemical reactivities (biological activities) of the title compounds, a gas-phase density functional theory (DFT) functional study was conducted using B3LYP. The B3LYP-optimized geometries are depicted in Figure 4 and Supplementary Materials for reference. As depicted in Figure 4, the individual bond length is as expected, which shows that the chosen theoretical level should be reliable. Moreover, the two C=O groups in both 1 and 2 have a difference in the bond length. The difference in the two C=O bond lengths was 0.023 Å, and 0.019 Å for 1, and 2, respectively. The converged geometry of the title compound, which was represented by the Cartesian coordinates of the respective atoms, was provided as the supporting information.
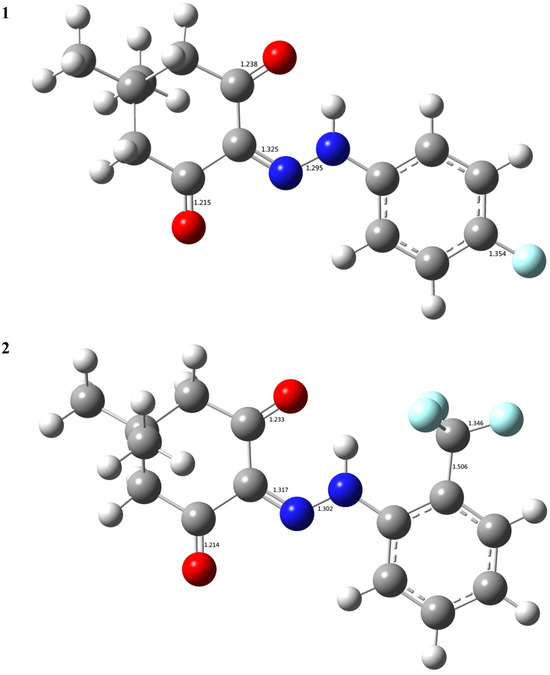
Figure 4.
The B3LYP-optimized geometries of the title compounds (the bond lengths in Å).
The pivotal aspect of the frontier orbital theory lies in its emphasis on the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO). Instead of solely contemplating the total electron density within the nucleophile, it is more instructive to focus on the electron density distribution within a molecule’s HOMO. Electrons in the HOMO exhibit the highest probability of engaging in nucleophilic attacks. Conversely, the electron density distribution in the LUMO serves as a reliable indicator for characterizing electrophilicity. Therefore, the frontier orbitals of the title compounds were further investigated in this study. As depicted in Figure 5, the frontier orbital of 1 or 2 was the linear combination of the π or π* orbital of the π-bonded moieties within 1 or 2.
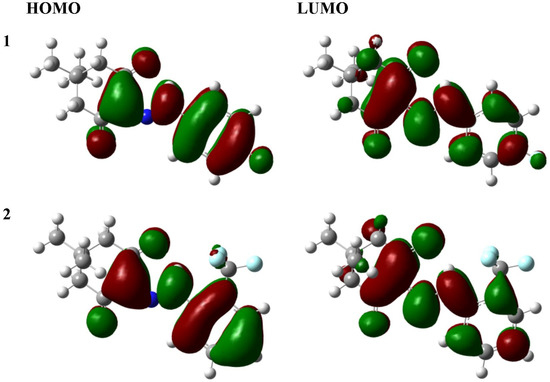
Figure 5.
The frontier orbital of the title compounds (the isovalue = 0.02 a.u.).
Molecular electrostatic potentials (MEPs) serve as a fundamental metric for gauging the strength of interactions among nearby charges, nuclei, and electrons at specific positions. This enables the examination of charge distribution and associated molecular properties. To enhance the interpretability of electrostatic potential information, a visual representation utilizing different colors was employed. Generally, regions in red signify the lowest electrostatic potential, rendering them susceptible to electrophilic attacks. Conversely, regions in blue indicate the highest electrostatic potential, making them susceptible to nucleophilic attacks. In this investigation, the total density matrix was utilized to derive the total density of the title compounds, and the resultant MEP was mapped onto their surfaces. As illustrated in Figure 6, oxygen atoms in the title compounds were identified as the sites prone to nucleophilic attack. Despite fluorine atoms possessing higher electronegativity than oxygen, those in both compounds 1 and 2 did not reside in the reddest region, where the MEP is not particularly negative. This can be elucidated by acknowledging that contributions to the molecules’ MEP arise from both electrons and nuclei.
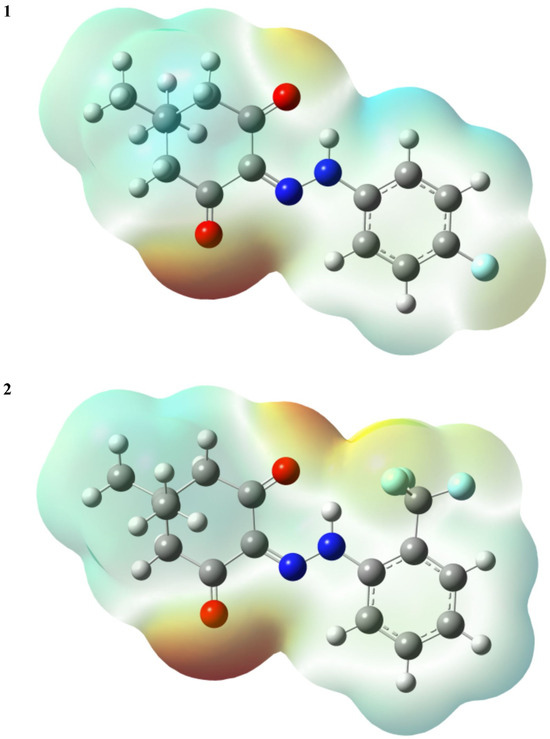
Figure 6.
The MEPs of the title compounds (the isovalue = 0.0004 a.u.).
3.3. Hirshfeld Surface Analysis
The Hirshfeld surfaces of the title compounds at standard resolution are illustrated in Figure 7. Transparency was applied to these surfaces to enable the visualization of the molecular moieties in a consistent orientation across all structures for which they were computed. The 3D dnorm surfaces serve to identify closely positioned intermolecular interactions. Negative (positive) values indicate that the intermolecular contacts are shorter (longer) than the van der Waals radii. The dnorm values are color-coded in red, white, or blue. Red regions signify closer contacts with negative dnorm values, whereas blue regions indicate longer contacts with positive dnorm values. Additionally, white areas represent contacts at the van der Waals separation with a zero value.
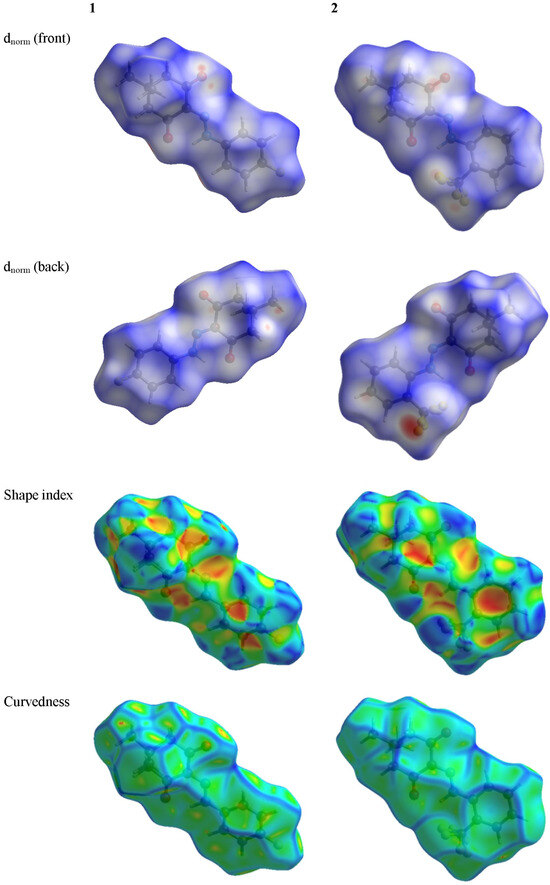
Figure 7.
The Hirshfeld surface of the title compounds.
Comparing the shape index and curvedness of 1 with those of 2, we can find that the π-π stacking interaction should be stronger in crystalline 2 than crystalline 1.
The 2D fingerprint plots elucidate the interaction between the specific atom pairs and facilitate the dissection of the overall fingerprint into contributions from various interaction types. Employing a standard view ranging from 0.6 to 2.4, with the de and di distance scales depicted on the graph axes and incorporating reciprocal contacts (Figure 8), it was observed that the most noteworthy interaction involving hydrogen in both title compounds was the H···H contact. A comparison between the 2D fingerprint plots of 1 and those of 2 revealed that the H···F contact exhibited a more substantial contribution to the intra- or intermolecular interactions in 2 compared to those in 1.
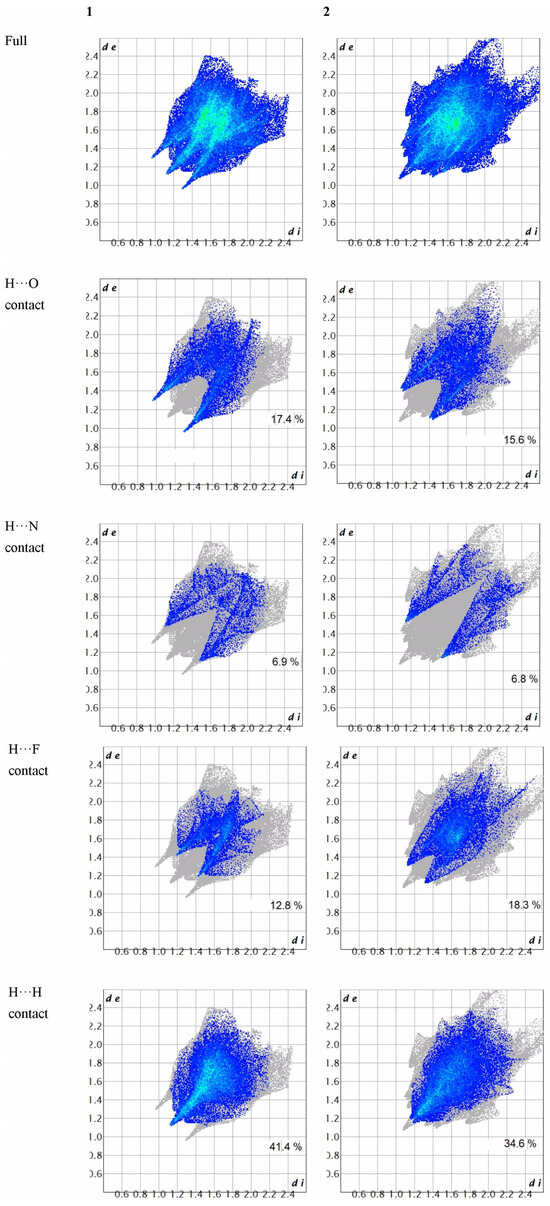
Figure 8.
The 2D fingerprint plots of the title compounds.
3.4. Energy Framework Analysis
Energy frameworks were constructed based on interaction types (electrostatic, polarization, dispersion, and exchange-repulsion) utilizing density functional theory (CE-B3LYP) in conjunction with the 6-31G(d,p) basis set. A cluster of molecules within a 3.8 Å radius was formed and considered around the central molecule, after which energy calculations were conducted. The interaction energy can be partitioned into
where k represents the scale factor, E_ele = electrostatic component, E_pol = polarization energy, E_dis = dispersion energy and E_rep = exchange-repulsion energy. As indicated in Table 1 and Table 2, the results demonstrate that the dispersion interaction significantly influences the interaction between the central molecule and its adjacent molecules in both crystalline 1 and 2. It is noteworthy that, despite the scale factor of the electrostatic component (k_elec) being the largest among all scale factors for both 1 and 2 (as detailed in Table 3), the dispersion interaction plays a predominant role. These interactions are visually depicted in Figure 9.

Table 1.
The energy framework of 1. (energy in kJ/mol and R in Å).
Table 2.
The energy framework of 2. (energy in kJ/mol and R in Å).
Table 3.
The scale factor for CE-B3YLP model energies with B3LYP/6-31G(d,p) monomer electron densities.
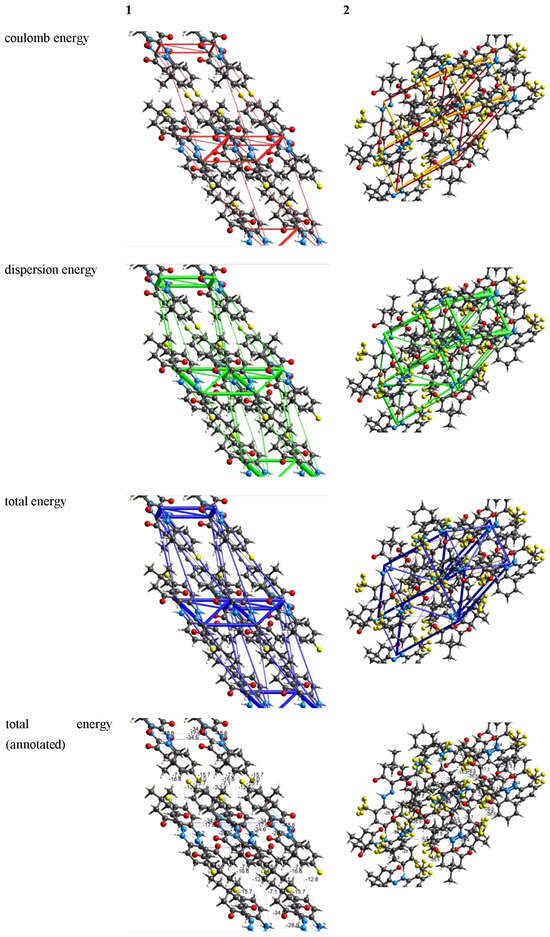
Figure 9.
The energy framework of the title compounds (tube size: 60, and cutoff: 5.00 kJ/mol).
3.5. The Molecular Docking Study
As mentioned earlier, β-diketones have been widely used as biologically active compounds, and then chosen to study and emphasize their possible target. According to the predicted result of the SwissTargetPrediction [50], the most probable target for 1 is COX-2. Hence, the anti-COX-2 activities of 1 and 2 were studied by the molecular docking in this study. IBF was taken as the reference to investigate the anti-COX-2 activities of the titled chemicals. Therefore, the .cif file of the IBF was downloaded from the PDB and converted into a .mol2 file by the gOpenBabel program [40]. Then, the MM2 method implemented in the Chem3D program was used to minimize the structure of IBF. Afterward, the geometry-optimization result of IBF was also saved as a .mol2 file. COX-2 (Cyclooxygenase-2) is a crucial enzyme involved in pain medication. When tissues are injured or inflamed, COX-2 is upregulated. It catalyzes the conversion of arachidonic acid into prostaglandins, particularly PGE2. Prostaglandins are lipid molecules that act as local messengers, sensitizing nerve endings to pain signals and promoting inflammation. PGE2 lowers the pain threshold by stimulating pain receptors (nociceptors) and increasing blood flow to the injured area. This amplifies pain perception and triggers the inflammatory response. Blocking COX-2 with specific inhibitors like NSAIDs (nonsteroidal anti-inflammatory drugs) reduces prostaglandin production, ultimately alleviating pain and inflammation.
Thus, for the synthesized derivatives, we aimed to analyse the possibility of bindings of compounds 1 and 2 toward COX-2 (PDB ID:4PH9; Resolution: 1.81 Å; R-Value Observed: 0.162). Before starting the actual docking, we validated the entire protocol with Glide, Schrodinger, LLC, NY, 2023 and the concern value of RMSD was obtained as 0.50 Å for re-docking the co-crystallized ligand, IBF. As depicted in Figure 10C, compound 1 had the majority of the hydrophobic interactions with amino acid residues such as Val117, Leu532, Leu360, Tyr356, Val350, Leu353, and Val524. It is also worth mentioning that we noticed one amide-Pi stacking with amino acid residue Gly527. Conversely, compound 2 had hydrophobic interactions with Val117, Leu532, Leu360, Leu353, and Tyr356 (Figure 10D). Moreover, the fluorine atom had interacted with Tyr386 via H-bonding. The amide-Pi interacting residue, i.e., Gly527 was retained for compound 2 as similar to 1. It is noticeable that the appearance of H-bond for compound 2 led the enhanced binding of COX-2, as evident from docking scores (Glide Dock Score, XP: –9.36 and –9.79 kcal/mol, respectively).
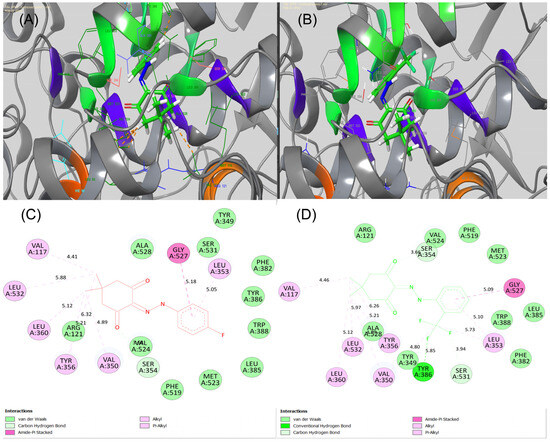
Figure 10.
(A,B) 3D-binding poses and (C,D) 2D-binding poses for Compounds 1 and 2 against the active site pocket of COX-2 (PDB code 4PH9), respectively. This figure was created by using Discovery Studio Visualizer, 2023.
The standard IBF, however, showed interactions with amino acid residues like Arg121, Val117, and Tyr 356 via H-bonding (Figure 11) (Glide Dock Score, XP: -7.98 kcal/mol). Other hydrophobic interactions were also seen for Leu360, val524, Val350, and Ala528 (alkyl or π-π interactions).
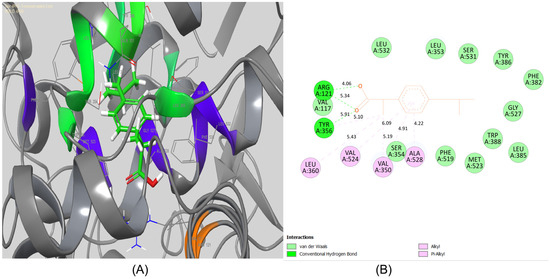
Figure 11.
(A) 3D binding pose and (B) 2D-binding pose for compound IBF against the active site pocket of COX-2 (PDB code 4PH9). This figure was created by using Discovery Studio Visualizer, 2023.
Notably, the Autodock 4 and Autodock vina were both performed via AMDock program [39], in which the pH was set to be 7.4 and the other docking parameters were set automatically by the used program. Table 4 listed the comparison of the anti-COX-2 activities of the titled compounds and IBF obtaining by the iGemDock program [34,35]. Table 4 shows that both 1 and 2 have better anti-COX-2 activity than the chosen reference. Furthermore, the activity of 1 was larger than that of 2, which may be due to the fact 1 could form stronger hydrogen bonding with COX-2 than 2 could. Both of the Autodock 4 and Autodock vina docking results were summarized as Table 5 showed. Similar to the iGemDock results, both the Autodock 4 and Autodock vina results also have shown that 1 and 2 have larger anti-COX-2 activity than the reference, IBF. However, the Autodock vina results showed that 2 should have a larger activity than 1, which disagreed with the iGemDock results.
Table 4.
The iGemDock docking results.
Table 5.
The docking results using the AMDock program.
3.6. Molecular Dynamics Analysis
We assessed the stability of both-docked ligands, denoted as COX-2:1 and COX-2:2, using a 150 ns molecular dynamics simulation (MD) carried out with the “Desmond” software by Schrodinger, LLC in 2023 (refer to Figure 12 and Figure 13). The MD system comprised a combined total of 58,768 atoms for COX-2:1 and 57,055 atoms for COX-2:2, inclusive of 16,624 and 16,022 water molecules, respectively. In the case of the COX-2:1 complex, 553 residues with 8859 atoms were engaged, while for COX-2:2, the involvement comprised 553 residues with 8858 atoms. In our dynamic simulations, we gauged the average atomic displacements over a specific time frame, employing the “RMSD” (Root Mean Square Deviation) parameter. Our RMSD analysis consistently revealed stable conformations. When ligands 1 and 2 were bound to the target 4PH9, the Cα-RMSD backbone values remained below 2.8 Å and 2.0 Å, respectively (refer to Figure 12A,B and Figure 13A,B). Additionally, the “Lig_fit_Prot” values consistently stayed below 2.5 Å for both simulations (see Figure 12B and Figure 13B), affirming the stability of the entire complexes over the 0–150 ns duration. We delved into local protein chain variations using the “RMSF” (Root Mean Square Fluctuation) plot (Figure 12B and Figure 13B), showing minor fluctuations in selected proteins without significant changes, highlighting the expected flexibility of these residues. Notably, we observed fewer fluctuations in amino acid residues for COX-2:2 compared to COX-2:1. Examining the “protein–ligand” interaction plot (Figure 12C), we discovered significant interactions, including hydrophobic interactions with amino acid residues Val117, Val350, Leu353, Tyr356, Leu360, Leu385, Tyr386, Phe519, and Leu532, but no ionic interactions (Figure 12F) in the complex COX-2:1. Water bridges formed with His90, Arg121, Leu353, Ser354, and Val524 for compound 1. Amino acid residues Gly527 and Ser531 established H-bonds. Similarly, the “protein–ligand” interaction plot (Figure 13F) for compound 2 displayed significant interactions, including hydrophobic interactions with amino acid residues Val117, Val350, Leu353, Tyr356, Leu360, Leu385, Tyr386, Phe519, and Leu532, with no ionic interactions. Water bridges were observed with His90, Arg121, Leu353, Ser354, and Val524.
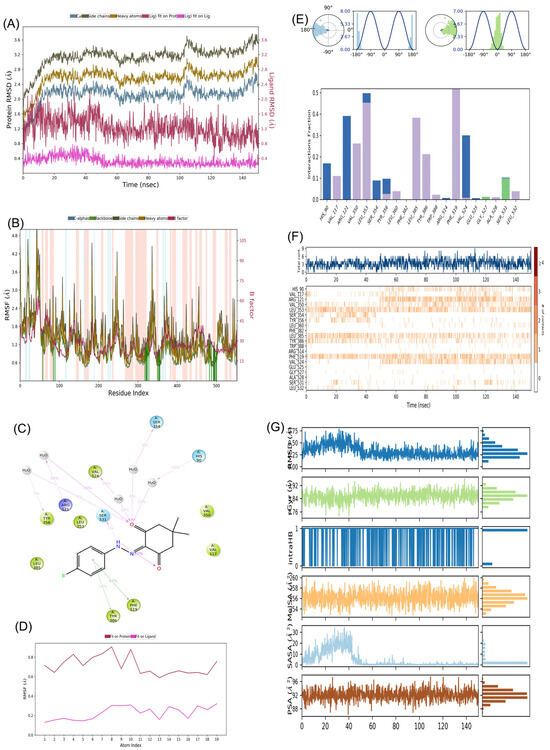
Figure 12.
MD simulation analysis for complex COX-2 (PDB code 4PH9):1 (A) RMSD plot; (B) RMSF analysis; (C) Ligand–Protein contact plot; (D) Ligand RMSF; (E) Ligand torsion profile; (F) Protein–Ligand interaction plot; (G) a timeline representation plot representing such interactions with amino acid residues over the simulation period of 150 ns for compound 1.
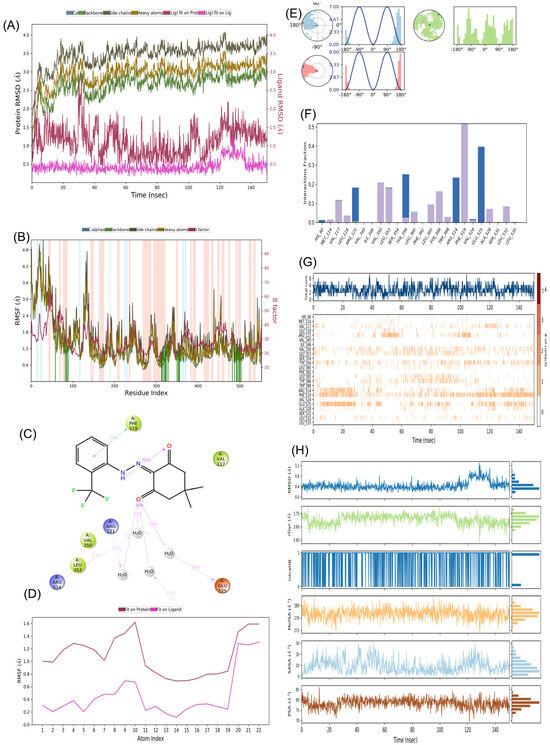
Figure 13.
MD simulation analysis for complex COX-2 (PDB code 4PH9):2 (A) RMSD plot; (B) RMSF analysis; (C) Ligand–Protein contact plot; (D) Ligand RMSF; (E) Ligand torsion profile; (F) Protein–Ligand interaction plot; (G) a timeline representation plot representing such interactions with amino acid residues over the simulation period of 150 ns and (H) Ligand properties for compound 2.
Figure 12G and Figure 13G presents a timeline representation of these interactions with amino acid residues over the 150 ns simulation period.
Figure 12C presents a “ligand–protein” contact plot, highlighting interactions that persisted for over 5.0% of the simulation time within the selected trajectory (0.00–150.00 ns) for compound 1. Notable amino acid residues included Tyr356 (5% H-bonding), Arg121 (39% H-bond), Ser531 (5% H-bond), Phe519 (43% pi–pi interactions), and Tyr386 (16% pi–pi interactions). Similarly, Figure 13C illustrates a “ligand–protein” contact plot for compound 2, revealing interactions lasting more than 5.0% of the simulation time in the same trajectory. Compound 2 had interacted with two key amino acids, as Glu525 and Tyr356 via H-bonding. Furthermore, the “ligand torsions plot” (Figure 12E and Figure 13E) succinctly summarizes conformational changes in each rotatable bond (RB) within the ligand over the entire simulation duration (0.00–150.00 ns) for 1 and 2, respectively. In conclusion, our results substantiate the stable conformations of the ligand–protein complexes over the 150 ns simulation period. The ligand RMSF plot for 2 (Figure 13D) had more fit to the protein site compared to 1 (Figure 12D), suggesting better binding of 2 toward COX-2.
3.7. The ADMET Study
The drug-likeness including the Lipinski’s parameters and pharmacokinetic properties of the titled compounds [51] were investigated by using the SwissADME online server. After analyzing 90% of orally active drugs, Lipinski reported that the drugs that reached phase II clinical testing have molecular weights ≤ 500 g/mol, number of hydrogen-bond donors (no. of H-bond donors) ≤ 5, and number of hydrogen-bond acceptors (no. of H-bond acceptors) ≤ 10 [51]. The SwissADME plot predicts additional physicochemical parameters associated with drug-likeness such as lipophilicity, topological polar surface area, solubility, and saturation [52,53,54,55,56,57,58,59,60,61]. The results could be summarized as shown in Figure 14 and Table 6. The SwissADME plot of the drug-likeness of the title compounds showed that all the physicochemical properties of the compounds were within the desirable range. The predicted pharmacokinetics showed that the skin permeation (logKp) was −6.08 cm/s, and −5.83 cm/s for 1, and 2, respectively. The more negative logKp of 1 indicated that 1 was less skin permeant than 2 [61].
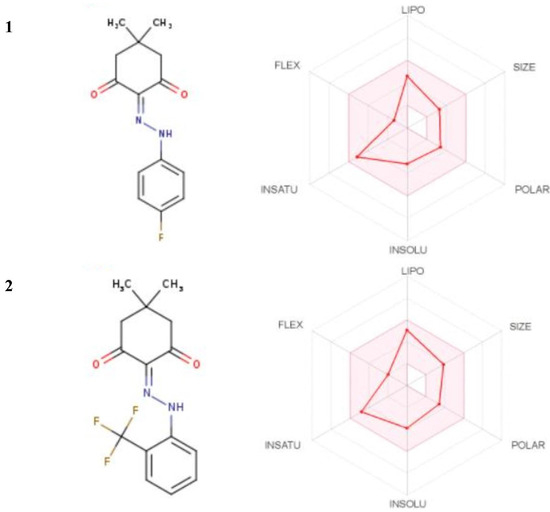
Figure 14.
The SwissADME plots of drug-likeness of the title compounds. The pink area represents the optimal range for each property (lipophilicity: XLOGP3 between −0.7 and +5.0, size: MW between 150 and 500 g/mol, polarity: TPSA between 20 and 130 Å2, solubility: logS not higher than 6, saturation: fraction of carbons in the sp3 hybridization not less than 0.25, and flexibility: no more than 9 rotatable bonds).
Table 6.
Summary of SwissADME predicted physicochemical descriptors and ADMET parameter of the titled compounds.
4. Conclusions
In this study, two following β-diketone derivatives 2-(2-(4-fluorophenyl)hydrazono)-5,5-dimethylcyclohexane-1,3-dione, and 5,5-dimethyl-2-(2-(2-(trifluoromethyl)phenyl)hydrazono)cyclohexane-1,3-dione were synthesized and characterized spectroscopically. Since β-diketones have been widely used as biologically active compounds, we studied them through molecular docking, molecular dynamics and the DFT as powerful techniques to investigate and evaluate the biological activity of our compounds against COX-2. Then, we analyzed ADMET further by the SwissADME plots and discovered that our compounds are well adsorbed and neither toxic nor carcinogenic. Our structures were also optimized through Hirschfeld surface analysis and energy framework, using the Crystal Explorer program. According to the results of the molecular docking studies, the title chemicals might have larger anti-COX-2 activities than IBF. However, since the predicted relative activity results of Glide, iGemdock, and Autodock vina were opposite, we were convinced that the in vitro anti-COX-2 activities of β-diketone derivatives are worth further study. To have more clarity, the MD study pointed out that compound 2 had better binding toward COX-2 and also obtained the best docked candidate from the Glide calculations. Furthermore, we firmly believe that the results of this paper should enlighten a broad spectrum in the research field of developing NSAIDs as COX-2 inhibitors.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/bioengineering10121361/s1, DFT-converged geometries, checkCIF files of the titled chemicals, and the whole amino acid sequences of the COX-1 and COX-2 enzymes.
Author Contributions
Conceptualization, M.M.K. and C.-H.L.; methodology, S.N.M., Y.E.B. and C.-H.L.; software, S.N.M. and C.-H.L.; validation, M.M.K., R.A.-S. and Y.E.B.; formal analysis, A.M.M. and A.Z.S.; investigation, A.M.M. and A.Z.S.; resources, Y.E.B.; data curation, R.A.-S., Y.E.B. and C.-H.L.; writing—original draft preparation, C.-H.L.; writing—review and editing, F.Z.G. and C.-H.L.; visualization, S.N.M. and C.-H.L.; supervision, M.M.K. and F.Z.G.; project administration, R.A.-S.; funding acquisition, R.A.-S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Researchers Supporting Project No. RSP-2023R353, King Saud University, Riyadh, Saudi Arabia.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available on request due to restrictions e.g., privacy or ethical.
Acknowledgments
The authors extend their appreciation to the Researchers Supporting Project, King Saud University, Riyadh, Saudi Arabia, for funding this work through grant No. RSP-2023R353. We also thank the National Center for High-Performance Computing (Taiwan) for providing computing time to do the DFT calculations. Special thanks go to the reviewers for their valuable suggestions or com-ments that have improved the quality of our manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vaidya, S.R.; Shelke, V.A.; Jadhav, S.M.; Shankarwar, S.G.; Chondhekar, T.K. Synthesis and Characterization of β-Diketone Ligands and Their Antimicrobial Activity. Arch. Appl. Sci. Res. 2012, 4, 1839–1843. [Google Scholar]
- Annamalai, S.; Balasubramaniyam, A.P.; Sabeta, K.; Rajnikant, V. Synthesis, spectral and RAHB studies on some arylhydrazones of β-Diketones: Crystal and molecular structures of 2-(2-(3-pyridyl)hydrazono)-5,5-dimethylcyclohexane-1,3-dione and 2-(2-(2-methoxyphenyl)hydrazono)-5,5-dimethylcyclohexane-1,3-dione. Struct. Chem. 2011, 22, 23–33. [Google Scholar]
- Jadhav, S.M.; Shelke, V.A.; Munde, A.S.; Shankarwar, S.G.; Patharkar, V.R.; Chondhekar, T.K. Synthesis, characterization, potentiometry, and antimicrobial studies of transition metal complexes of a tridentate ligand. J. Coord. Chem. 2010, 23, 4153–4164. [Google Scholar] [CrossRef]
- Karvembu, R.; Jayabalakrishnan, C.; Natarajan, K. Thiobis(β-diketonato)-bridged binuclear ruthenium(III) complexes containing triphenylphosphine or triphenylarsine. Synthetic, spectral, catalytic and antimicrobial studies. Transit. Met. Chem. 2002, 27, 574–579. [Google Scholar] [CrossRef]
- Ryszard, G.; Erkki, K.; Henryk, J.; Reijo, K.; Maija, N.; Borys, O. Predominance of 2-arylhydrazones of 1,3-diphenylpropane-1,2,3-trione over its proton-transfer products. J. Phys. Org. Chem. 2001, 14, 797–803. [Google Scholar]
- Lasri, J.; Gajewski, G.; Guedes da Silva, M.; Fátima, C.; Kuznetsov, M.L.; Fernandes, R.; Pombeiro Armando, J.L. Solvent-dependent reactivities of acyclic nitrones with β-diketones: Catalyst-free syntheses of endiones and enones. Tetrahedron 2012, 68, 7019–7027. [Google Scholar] [CrossRef]
- Kumar, C.U.; Sethukumar, A.; Prakasam, B. Arul. Synthesis and spectral studies of some 4H-pyran derivatives: Crystal and molecular structure of isobutyl 6-amino-5-cyano-2-methyl-4-phenyl-4H-pyran-3-carboxylate. J. Mol. Struct. 2013, 1036, 257–266. [Google Scholar] [CrossRef]
- Lingaiah, B.P.V.; Reddy, G.; Venkat; Yakaiah, T.; Narsaiah, B.; Reddy, S.N.; Yadla, R.; Rao, P. Shanthan. Efficient and Convenient Method for the Synthesis of Poly Functionalised 4H-Pyrans. Synth. Commun. 2004, 34, 4431–4437. [Google Scholar] [CrossRef]
- Gou, S.-B.; Wang, S.-X.; Li, J.-T. D,L-Proline-Catalyzed One-Pot Synthesis of Pyrans and Pyrano [2,3-c]pyrazole Derivatives by a Grinding Method under Solvent-Free Conditions. Synth. Commun. 2007, 37, 2111–2120. [Google Scholar]
- John, V.D.; Krishanankutty, K. Antitumour activity of synthetic curcuminoid analogues (1,7-diaryl-1,6-heptadiene-3,5-diones) and their copper complexes. Appl. Organomet. Chem. 2006, 20, 477–482. [Google Scholar] [CrossRef]
- Pihlaja, K.; Taskinen, A.; Gawinecki, R.; Janota, H. Behaviour of 1,3-diphenyl-2-arylhydrazono-1,3-propanediones under electron ionisation. Rapid Commun. Mass Spectrom. 2003, 17, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Maurya, R.C.; Rajput, S. Oxovanadium(IV) complexes of bioinorganic and medicinal relevance: Synthesis, characterization, and 3D molecular modeling and analysis of some oxovanadium(IV) complexes involving O,O-donor environment. J. Mol. Struct. 2004, 687, 35–44. [Google Scholar] [CrossRef]
- Hinckley, C.C. Paramagnetic shifts in solutions of cholesterol and the dipyridine adduct of trisdipivalomethanatoeuropium(III). A shift reagent. J. Am. Chem. Soc. 1969, 91, 5160–5162. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, B.; Buono-Core, G.E. Photochemical properties of 1,3-diketonate transition metal chelates. J. Photochem. Photobiol. A 1990, 52, 1–25. [Google Scholar] [CrossRef]
- Legan, M. Cyclooxygenase-2, p53 and glucose transporter-1 as predictors of malignancy in the development of gallbladder carcinomas. Bosn, J. Basic Med. Sci. 2010, 10, 192–196. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Menter, D.G.; Schilsky, R.L.; DuBois, R.N. Cyclooxygenase-2 and cancer treatment: Understanding the risk should be worth the reward. Clin. Cancer Res. 2010, 16, 1384–1390. [Google Scholar] [CrossRef]
- Wang, D.; Patel, V.V.; Ricciotti, E.; Zhou, R.; Levin, M.D.; Gao, E.; Yu, Z.; Ferrari, V.A.; Lu, M.M.; Xu, J.; et al. Cardiomyocyte cyclooxygenase-2 influences cardiac rhythm and function. Proc. Natl. Acad. Sci. USA. 2009, 106, 7548–7852. [Google Scholar] [CrossRef]
- Hawash, M.; Jaradat, N.; Abualhasan, M.; Şüküroğlu, M.K.; Mohammed, T.; Qaoud, M.T.; Kahraman, D.C.; Heba Daraghmeh, H.; Maslamani, L.; Mais Sawafta, M.; et al. Design, synthesis, molecular docking studies and biological evaluation of thiazole carboxamide derivatives as COX inhibitors. BMC Chem. 2023, 17, 11. [Google Scholar] [CrossRef]
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran J. Pharm. Res. 2011, 10, 655–683. [Google Scholar]
- Beck, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter. 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Mohammed Hawash, M.; Qaoud, M.T.; Jaradat, N.; Abdallah, S.; Issa, S.; Adnan, N.; Hoshya, M.; Sobuh, S.; Hawash, Z. Anticancer Activity of Thiophene Carboxamide Derivatives as CA-4 Biomimetics: Synthesis, Biological Potency, 3D Spheroid Model, and Molecular Dynamics Simulation. Biomimetics 2022, 7, 247. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16, Revision C.01/C.02; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Spackman, M.A.; Byrom, P.G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 215–220. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2004, 60, 627–668. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–28. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.M.A.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; University of Western Australia: Crawley, Australia.
- Salih, T. A Comparative Study for the Accuracy of Three Molecular Docking Programs Using HIV-1 Protease Inhibitors as a Model. Iraqi. J. Pharm. Sci. 2022, 31, 160–168. [Google Scholar] [CrossRef]
- Castro-Alvarez, A.; Costa, A.M.; Vilarrasa, J. The Performance of Several Docking Programs at Reproducing Protein-Macrolide-Like Crystal Structures. Molecules 2017, 22, 136. [Google Scholar] [CrossRef]
- Ivanova, L.; Karelson, M. The Impact of Software Used and the Type of Target Protein on Molecular Docking Accuracy. Molecules 2022, 27, 9041. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-M.; Chen, C.-C. GEMDOCK: A generic evolutionary method for molecular docking. Proteins Struct. Funct. Genet. 2004, 55, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.-C.; Chen, Y.-F.; Lin, S.-R.; Yang, J.-M. iGEMDOCK: A graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinform. 2011, 12, S33. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. AMDock: A versatile graphical tool for assisting molecular docking with Autodock Vina and Autodock4. Biol. Direct 2020, 15, 12. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Geoffrey, R.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, A.; Abouzied, A.S.; Alamri, A.; Anwar, S.; Ansari, M.; Khadra, I.; Zaki, Y.H.; Gomha, S.M. Synthesis, Molecular Docking, and Dynamic Simulation Targeting Main Protease (Mpro) of New, Thiazole Clubbed Pyridine Scaffolds as Potential COVID-19 Inhibitors. Curr. Issues Mol. Biol. 2023, 45, 1422–1442. [Google Scholar] [CrossRef] [PubMed]
- Mali, S.N.; Pandey, A. Synthesis of new hydrazones using a biodegradable catalyst, their biological evaluations and molecular modeling studies (Part-II). J. Comput. Biophys. Chem. 2022, 21, 857–882. [Google Scholar] [CrossRef]
- Ghosh, S.; Mali, S.N.; Bhowmick, D.N.; Pratap, A.P. Neem oil as natural pesticide: Pseudo ternary diagram and computational study. J. Indian Chem. Soc. 2021, 98, 100088. [Google Scholar] [CrossRef]
- Mali, S.N.; Sawant, S.; Chaudhari, H.K.; Mandewale, M.C. In silico appraisal, synthesis, antibacterial screening and DNA cleavage for 1, 2, 5-thiadiazole derivative. Curr. Comput. Aided. Drug Des. 2019, 15, 445–455. [Google Scholar] [CrossRef]
- Mali, S.N.; Pandey, A. Multiple QSAR and molecular modelling for identification of potent human adenovirus inhibitors. J. Indian Chem. Soc. 2021, 98, 100082. [Google Scholar] [CrossRef]
- Mali, S.N.; Pandey, A.; Bhandare, R.R.; Shaik, A.B. Identification of hydantoin based Decaprenylphosphoryl-β-d-Ribose Oxidase (DprE1) inhibitors as antimycobacterial agents using computational tools. Sci. Rep. 2022, 12, 16368. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of Octanol−Water Partition Coefficients by Guiding an Additive Model with Knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. Revisiting the General Solubility Equation: In Silico Prediction of Aqueous Solubility Incorporating the Effect of Topographical Polar Surface Area. J. Chem. Inf. Model. 2012, 52, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.S. ESOL: Estimating Aqueous Solubility Directly from Molecular Structure. J. Chem. Inf. Comput. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Ritchie, T.J.; Ertl, P.; Lewis, R. The graphical representation of ADME-related molecule properties for medicinal chemists. Drug Discov. Today 2011, 16, 65–72. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V.A. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Di, L. The role of drug metabolizing enzymes in clearance. Expert Opin. Drug Metab. Toxicol. 2014, 10, 379–393. [Google Scholar] [CrossRef]
- Potts, R.O.; Guy, R.H. Predicting Skin Permeability. Pharm. Res. 1992, 9, 663–669. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).