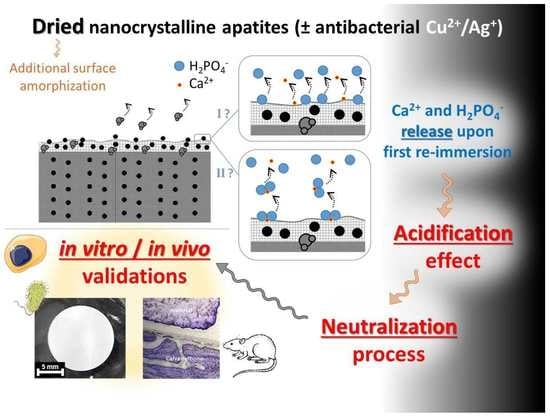
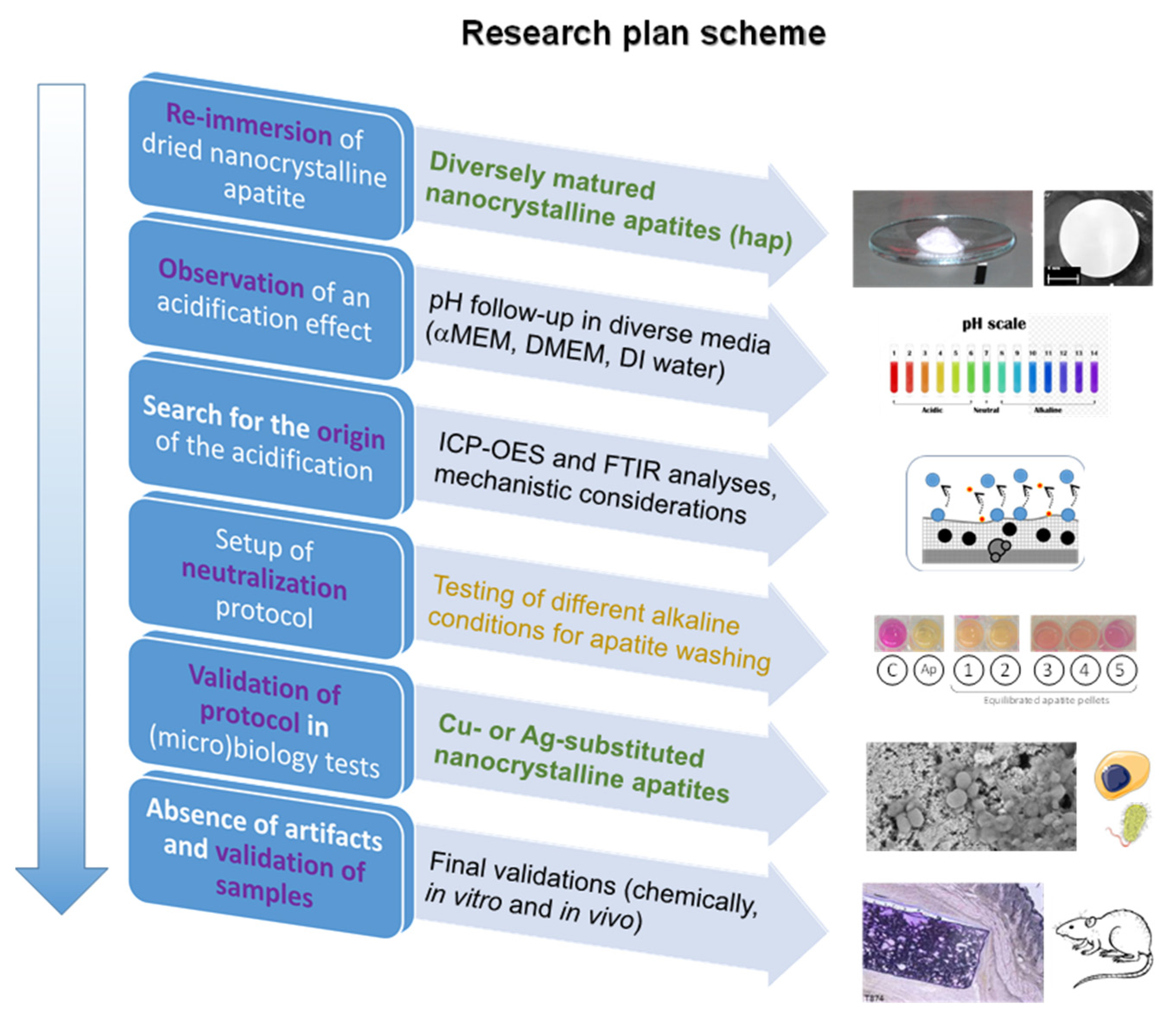
3.1. Occurrence and Origin of an Acidification Event after First Re-Immersion of Dried Biomimetic Apatites
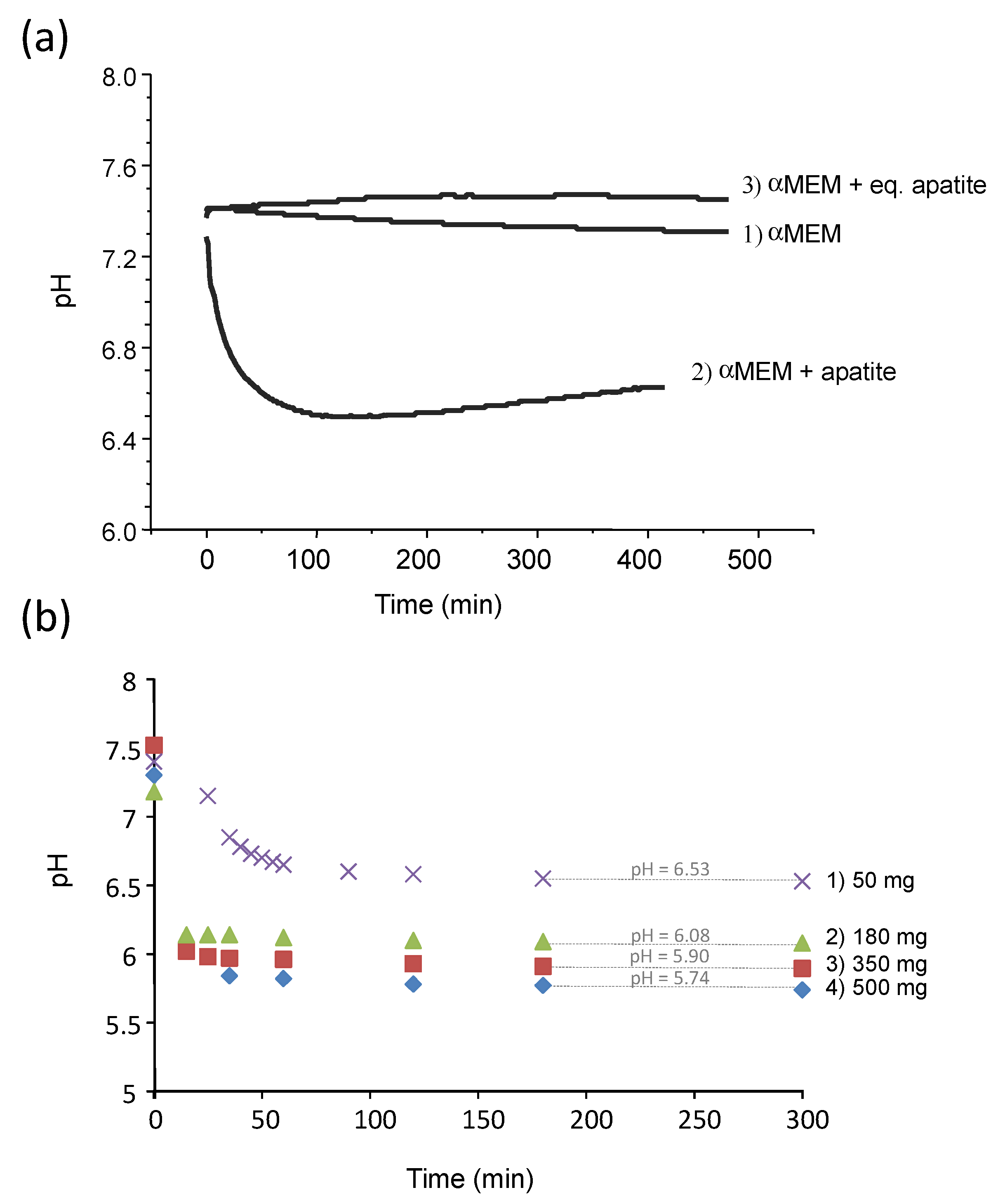
The pH of the pure αMEM medium was monitored over 7 h at 37 °C, showing a stable pH value in the range from 7.4 to 7.3 (
Figure 2a, curve 1), even in contact with atmospheric air (thus without the use of a cell culture incubator fed with CO
2). In contrast, in the presence of the freeze-dried (non-carbonated) biomimetic apatite hap-1d (
Figure 2a, curve 2) previously characterized in detail [
1], the pH of the medium dropped significantly from 7.4 down to about 6.6 within the same time frame. A similar effect was noticed in other media such as DMEM. These results highlight the existence of interactions between apatite nanocrystals and the surrounding solution, leading to an acidification effect. As mentioned above, these observations indicate other authors’ reports that have yet to be explained so far [
9]. In order to avoid any potential artefact due to a possible complexation process involving the ions and molecular species contained in αMEM, the immersion was reproduced in deionized water (pH set to 7.4 with NaOH/HCl). As in αMEM, a similar pH drop from 7.4 down to 6.53 was noticed again after immersing hap-1d (
Figure S1 (in Annex—Supplementary Information), curve a). It may be noted that the pH stabilization occurred significantly faster in pure water than in αMEM, which may reasonably be related to the presence of molecular entities in αMEM delaying its kinetic evolution toward equilibrium.
To further investigate this acidification phenomenon, pH was also followed by immersing a nanocrystalline apatite sample matured for only 20 min (hap-20 min), thus exhibiting a significantly lower degree of maturation and a larger amount of the hydrated layer on the nanocrystal surface, as explained previously [
1,
10]. The stabilized pH for hap-20 min (
Figure S1, curve b) then reached 6.41. This pH value refers to more acidic conditions than for hap-1d (all other conditions being the same). Additionally, the slope of the pH = f(t) curve was dramatically steepened for hap-20 min compared to hap-1d, as the stabilization was obtained after ~30 s for hap-20 min versus ~70 s for hap-1d. These findings indicate that the degree of acidification of the solution after re-immersion of the freeze-dried nanocrystalline apatites directly depends on their maturation state: the immersion of less matured samples (i.e., exhibiting a larger amount of the non-apatitic hydrated layer on the surface of the nanocrystals) generates a greater acidification effect.
The effect of the mass of dried apatite sample immersed in a constant volume (20 mL deionized water, initial pH 7.4) was also investigated for hap-1d (
Figure 2b). As may be noticed, a clear tendency of increased acidification (stabilized pH values from 6.53 down to 5.74) was found upon increasing the amount of immersed freeze-dried sample. This phenomenon cannot be attributed to a regular dissolution behavior of a solid phase since no obviously acidic entity constituted the sample composition and it must be explained by a more complex surface/solution interaction mechanism. In addition, this phenomenon is very rapid, as is initiated from the very first seconds following re-immersion in an aqueous medium.
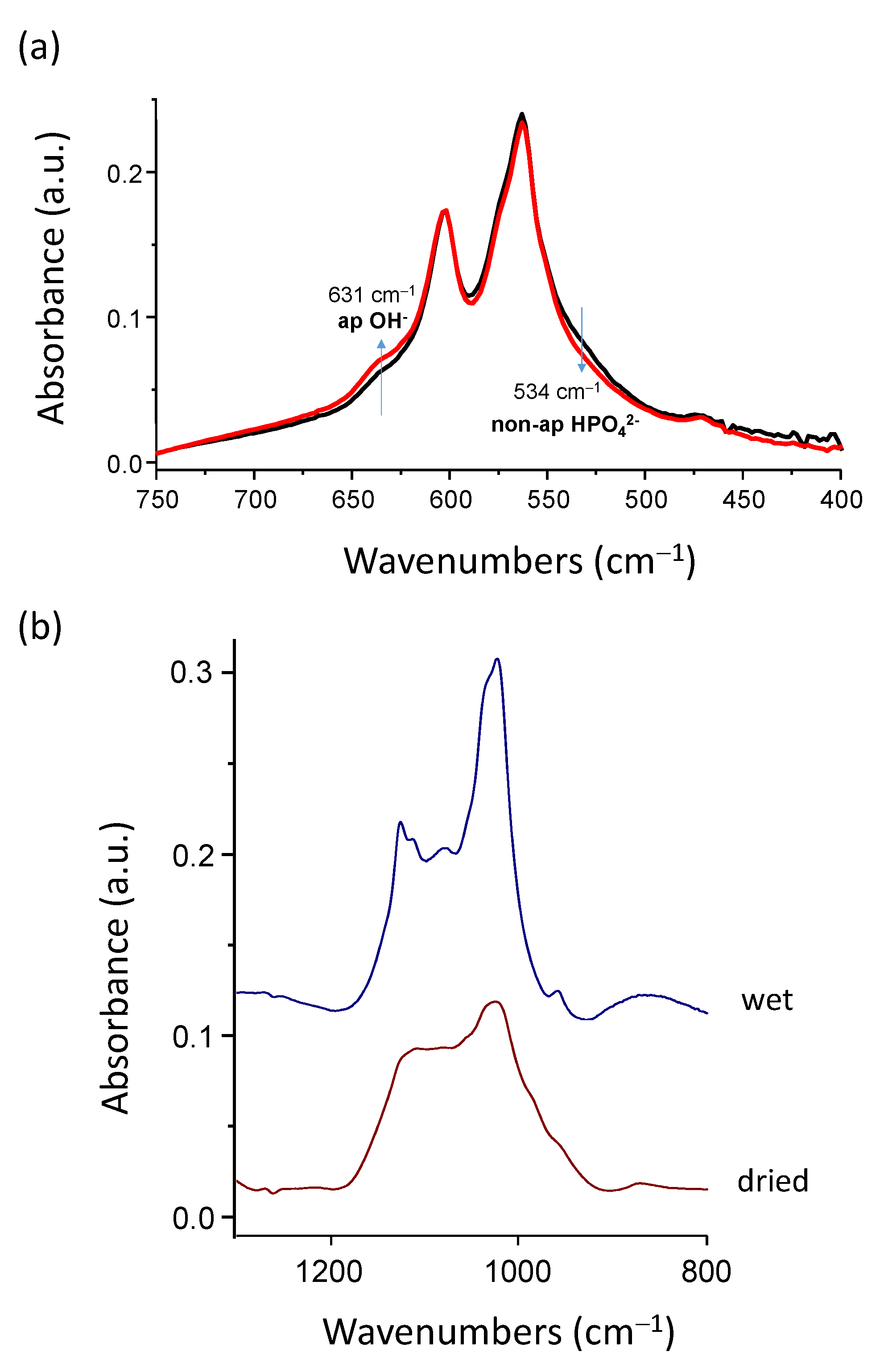
In order to explore the underlying mechanism of this acidification phenomenon, the FTIR analyses of freeze-dried hap-1d were carried out before and after sample immersion for ~150 min, which corresponds roughly to the stabilization delay of the pH drop (see
Figure 2). This rather short period of immersion is also interesting as it avoids any significant post-maturation; thus, we may assign any observed feature to the acidification process itself. In particular, the ν
4PO
4 vibration domain was analyzed, using the spectral decomposition procedure detailed previously [
1]. By comparison with the spectral features of the initial hap-1d freeze-dried powder, the FTIR results after re-immersion point out (
Figure 3a) a decrease in the proportion of non-apatitic HPO
42− ion content (band at ~534 cm
−1). This evolution cannot be assigned to post-maturation due to the very limited duration of immersion (150 min) as compared to the initial maturation time (1 day). The immersion process thus directly promotes a noticeable decrease in surface HPO
42− ions relative to other species. Since it is also accompanied by solution acidification, these results strongly suggest that some protonated phosphate ions are released into the solution at the time of re-immersion. Besides this decrease in non-apatitic HPO
42− ion content, we can also note that the proportion of the band at ~632 cm
−1 assignable to a libration band of apatitic OH
− ions also somewhat increases. This effect may again be explained by the release of some phosphate ions to the solution upon re-immersion, thus lowering the total amount of phosphate ions and, in turn, artificially increasing the relative proportion in the ν
4PO
4 domain of any other species active in IR, such as OH
−.
The non-apatitic surface layer on biomimetic apatite nanocrystals contains a significant amount of protonated “non-apatitic” HPO
42− phosphate ions [
1]. In contrast, “non-apatitic” PO
43− species are present only in a very limited amount at the surface of the nanocrystals [
1,
10]. In other words, the phosphate speciation at the surface is clearly favoring the protonated HPO
42− form over PO
43−. The above FTIR findings have pointed to the release of protonated phosphate ions upon first re-immersion. However, the pK
a of the acid-base couple H
2PO
4−/HPO
42− is 7.2, meaning it is, thus, very close to the physiological value. Therefore, at pH 7.4, the release of HPO
42− ions from the solid phase would tend to consume (rather than release) some H
+ from the solution to form a balanced amount of H
2PO
4−/HPO
42− species, which is not observed here as evidenced by the pH drop. A plausible mechanism explaining both the acidification effect and the loss of protonated phosphate species as detected using FTIR would instead be based on the release of diprotonated H
2PO
4− ions. Such ions could form upon interaction among adjacent HPO
42− ions, via proton displacement (hopping). Such a hopping scenario can be schematized using the following scheme:
With the H
2PO
4− ions formed at the solid top surface having two negative charges, they would become less attracted by surface Ca
2+ cations than HPO
42− (due to their less negative charge) and could then be more easily released upon re-immersion:
This release process is indeed in agreement with Christoffersen’s findings on apatite dissolution [
11], stating that “when a negative surface ion has reacted with a H
+ ion, all electrostatic bonds between that surface ion and the surrounding Ca
2+ ions are weakened, and the activation energy needed to remove the negative surface ion is thus reduced”.
By releasing diprotonated H
2PO
4− ions, an acidification effect would finally be expected due their tendency, when in solution in neutral conditions, to partly form HPO
42− using the acid-base reaction:
For neutrality reasons, the release of negative H2PO4− ions can only occur, however, if cations simultaneously leave the solid or if negative ions from the solution replace them. In pure water, the only conceivable option lies in the concomitant release of Ca2+ surface ions (one Ca2+ ion being released for two H2PO4− ions to preserve electroneutrality).
In order to validate this mechanistic scenario, the calcium and phosphate ions released in solution upon re-immersion in water at initial pH 7.4 of hap-20 min, hap-3 h, and hap-1d samples (50 mg) were titrated using ICP-OES. The results (
Table S1) confirmed our above hypothesis by evidencing the actual release of both calcium and phosphate ions and, as expected, the Ca/P ratios reached in solution were measured close to 0.50 (in the range 0.44–0.56). Therefore, these results strongly substantiate our hypothesis of the released dissolved species corresponding to the overall stoichiometry “Ca(H
2PO
4)
2“ upon the re-immersion of dried nanocrystalline apatite powders.
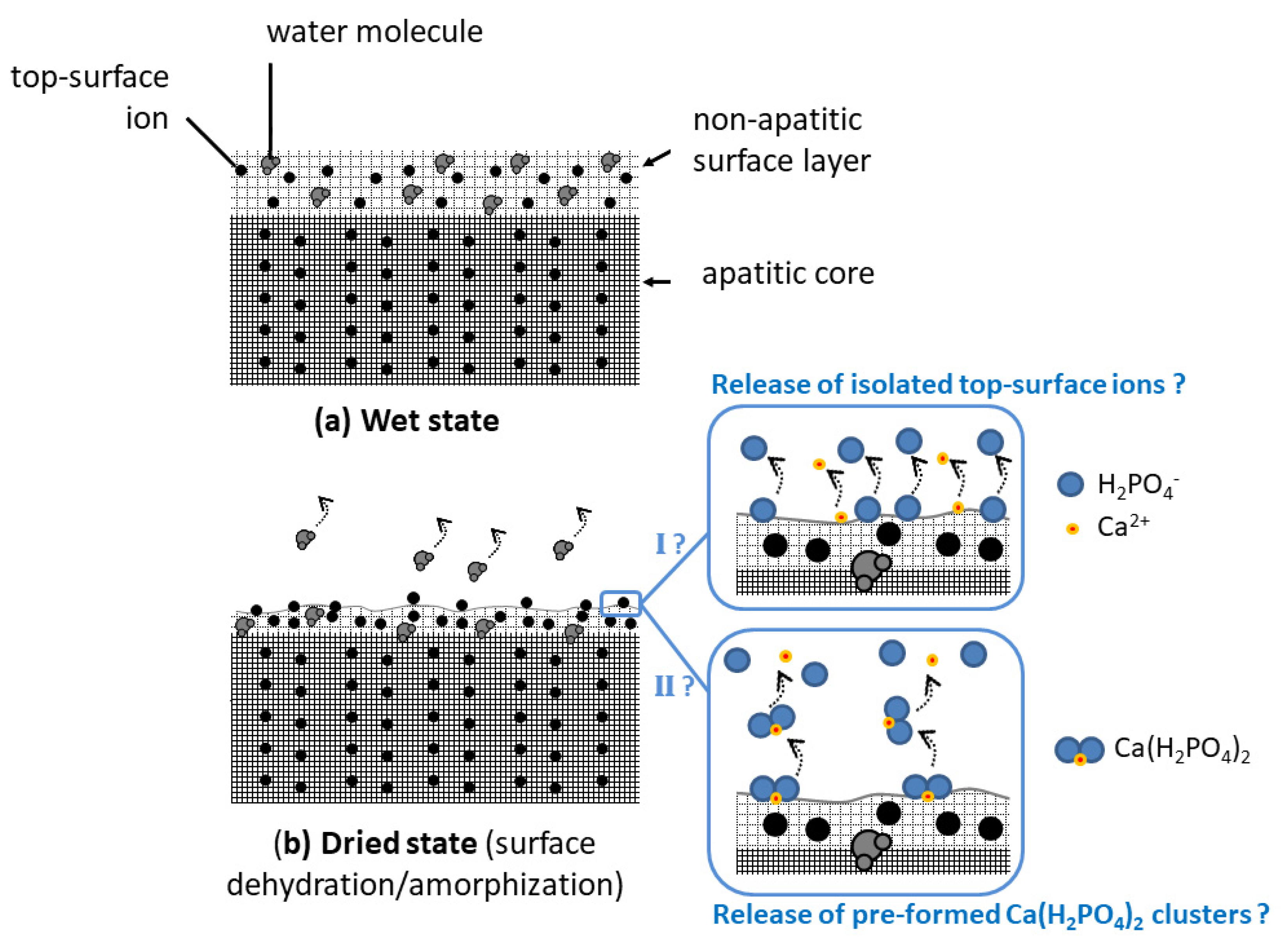
All the above results point out that, upon the re-immersion of dried nanocrystalline apatites, the fast release of ions corresponding to the global stoichiometry Ca(H2PO4)2. It is not clear at this point whether Ca(H2PO4)2 “clusters” may have formed/accumulated on the crystal top surface during the drying step itself or if the involved ions are simply concomitantly released in the solution upon re-immersion. In any case, the drying step is likely to partially dehydrate the utmost surface layer, which may disorganize the ionic environment of top-surface ions. This partial loss of water is indeed expected to modify the H-bonding array within the hydrated surface layer. Additionally, the (large) oxygen atoms from the H2O surface molecules may, in the wet state, participate in the coordination sphere of non-apatitic surface ions and the water loss could then destabilize the top-surface arrangement of the ions. Note that the formation of a separate monocalcium phosphate phase, such as monohydrate (MCPM) or anhydrous (MCPA), has never been noticed using common methods (XRD, FTIR, Raman, and NMR) including by us. Only top-surface ions are probably implicated, thus limiting the detection of eventual Ca(H2PO4)2 clusters at the crystals surface even if they exist as such on the dried surface. In both hypotheses anyway (cluster or concomitant ion release), the phosphate ions should be released in their diprotonated form H2PO4− to explain our observations (acidification, Ca/P ~0.5 in solution, and IR detection of a decrease in non-apatitic HPO42− content). This speciation is thought to arise from protons hopping from one HPO42− ion to the other, as described in Equation (1).
Our findings thus advocates that the acidification event observed after the first re-immersion of dried nanocrystalline apatites is linked to the alteration of the environment of top-surface ions from the surface layer. The re-immersion of other apatitic calcium phosphates where no hydrated layer is present on the crystal surface, such as stoichiometric hydroxyapatite (HA) Ca
10(PO
4)
6(OH)
2 or apatitic tricalcium phosphate (TCP
ap) Ca
9(PO
4)
5(HPO
4)(OH) (in which all HPO
42− ions are localized in apatitic chemical environments) was also followed for verification and, as anticipated, it did not lead to any significant pH change (
Figure S2). This observation confirms that the acidification process is assignable to the non-apatitic surface layer present on biomimetic apatite nanocrystals.
In order to further inspect the effect of drying on the nanocrystals physicochemical features, FTIR analyses were performed on wet, freshly precipitated apatite nanocrystals. The analysis of wet samples with a short maturation time such as 20 min clearly showed the existence of a fine band structure especially visible in the ν
3PO
4 domain (
Figure 3b). These fine features can be related to the specific organization of biomimetic apatite nanocrystals, exhibiting a non-apatitic hydrated ionic layer on their surface [
12]. Upon drying, however, this fine IR structure becomes noticeably altered, leading to a smoothened envelope (
Figure 3b). This loss of vibrational details can be explained by some additional surface “amorphization” due to the removal of some “structural” water molecules from the hydrated ionic layer. These observations favor the hypothesis of a partial denaturation of the ionic environment of utmost surface ions and they also agree with the easier release of surface ions upon re-immersion. This may be schematized as shown in
Figure 4, where the two hypothetical pathways discussed above are illustrated: pathway I supposing the release upon the first re-immersion of isolated top-surface ions and pathway II supposing the release of the pre-formed Ca(H
2PO
4)
2 clusters.
The above findings thus indicate that the (first) re-suspension of dried apatite nanocrystals in an aqueous medium is accompanied by an acidification phenomenon arising from the equilibration of the crystal’s surface with the solution, leading to an ultimate release of protons. The underlying mechanism is thought to originate from the protonated character of phosphate surface ions on apatite nanocrystals combined with top-surface alterations of ionic environments due to the additional amorphization effect generated by the drying process.
3.2. Circumventing this Acidification Effect
The above findings have pointed out the occurrence of an acidification effect upon re-immersion of dried nanocrystalline apatite samples: a mechanism was then established to explain all the experimental observations made (FTIR, ICP-OES, pH follow-up), based on the ultimate release of H2PO4− ions along with their calcium counter-ions. This effect may thus be linked to the presence of high amounts of protons on the non-apatitic surface layer on the nanocrystals, and thus to a marked acidic character. Based on this understanding, one way to circumvent this acidification could then lie in an equilibration step under alkaline conditions prior to drying.
In this view, the effect of different “equilibration” alkaline solutions during precipitate filtration was followed qualitatively by exposing each equilibrated apatite pellet to DMEM medium containing an internal pH indicator (phenol red), as summarized on
Figure 5. A clear color transition of the pH indicator was observed from pink (for pure neutral DMEM at a pH 7.4) to light yellow for the reference apatite pellet exempt of equilibration washing step, thus evidencing a clear acidification effect. For equilibration washing carried out in aqueous solutions of NaOH (pH 9 and 11), an intermediate orange coloration was observed, indicative of an improvement (limitation of the acidification effect) but not totally satisfactory in these conditions. Even better results were obtained through equilibration in Na
2HPO
4 pH 9 and Na3PO4 · 12H2O pH 10 which led in contrast to red tones, thus indicating close-to-physiological pH value. Finally, the least color variation compared to the control was noticed for equilibration washing in Na3PO4 · 12H2O pH 11 (followed by final washing with pure deionized water). This last equilibration solution clearly led to the best result, with only a slight clearing of the pH indicator initial pink coloration but no signs of acidification, upon immersion of equilibrated apatite pellet. Experiments were repeated in αMEM to follow quantitatively the pH of the solution over 7 h (added in
Figure 2a as curve 3). In this case, as may be expected from the results of
Figure 5, the pH value remained stable around 7.4, thus confirming the absence of acidification effect in this case.
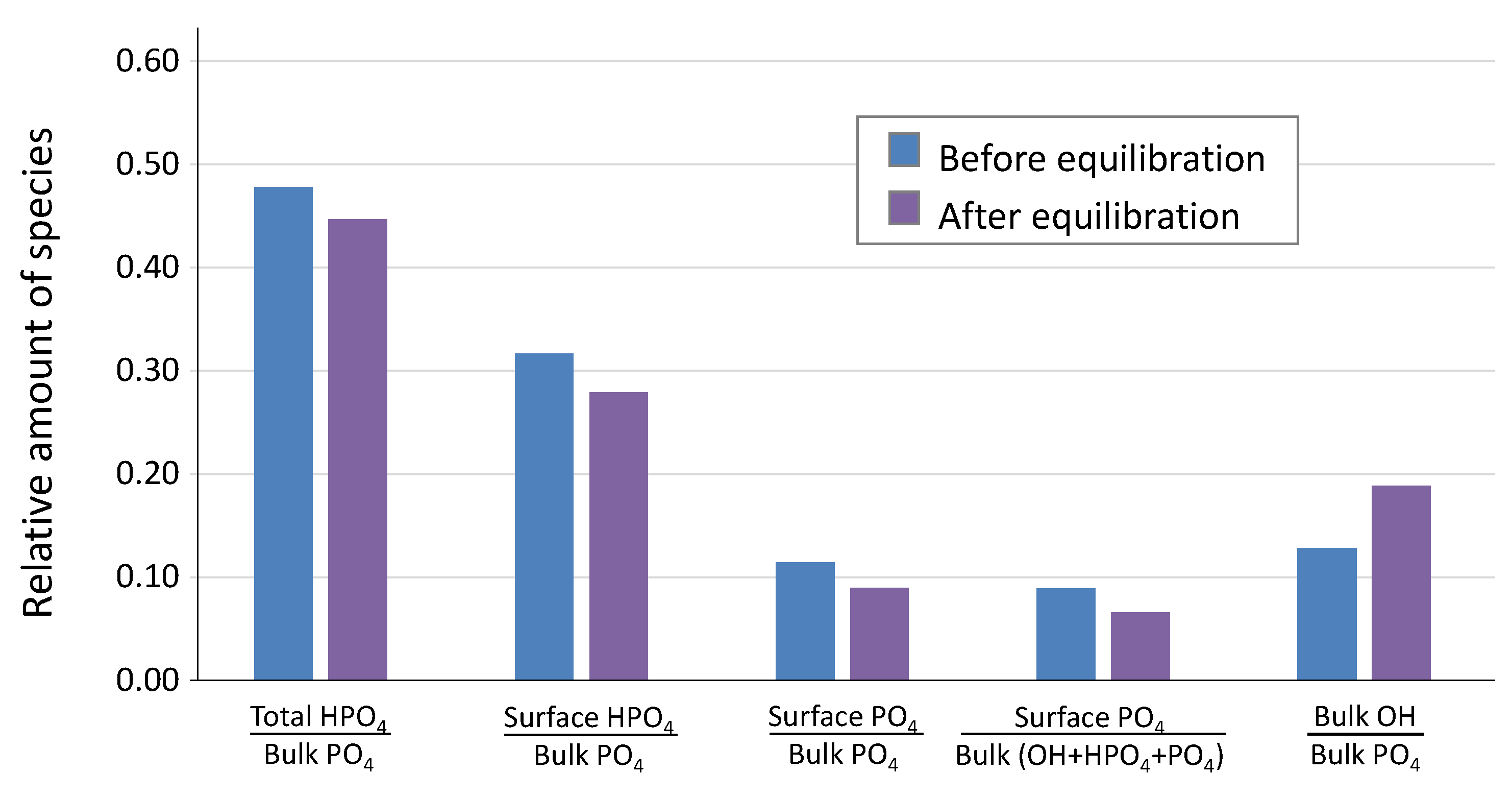
At this stage, it was interesting to investigate how the surface layer composition was modified by operating the equilibration step in the retained alkaline conditions. To this aim, FTIR decompositions in the ν
libOH-ν
4PO
4 domain were carried out before and after the equilibration/neutralization step (
Figure 6), following the spectral decomposition methodology already described [
1]. The proportion of the OH
− band appeared to increase in this spectral domain after the neutralization protocol, which is indicated by the increase in the [bulk OH/bulk PO
4] ratio. Taking into account the high alkaline pH of 11, and the high reactivity of the freshly prepared apatite gel during this washing stage, this observation points to the increased filling of apatitic channels by additional OH
− ions during the alkaline treatment. The high concentration in OH
−(aq) in the washing solution upon dissolution of the Na
3PO
4 · 12H
2O salt can be deduced from the following equation, by taking into account that the nearest pK
a value is that of the couple HPO
42−/PO
43− at 12.7, thus favoring the HPO
42− speciation at pH 11:
Additionally, the absence of acidification effect after re-immersion of dried apatite treated with this alkaline washing protocol strongly suggests that the density of surface protons (initially in the form of top-surface HPO
42− ions) has coincidently decreased, which is corroborated by the decrease in the [surface HPO
4/bulk PO
4] ratio seen in
Figure 6. A first simplistic view of this “surface neutralization” could initially be considered as follows:
where S represents the top-surface of the crystals in contact with the solution. However, both the additional inclusion of OH
− in the crystals apatitic channels and Equation (5) would lead to an excess of negative charges in the solid and thus of positive charges in the solution. In order to maintain the overall neutrality of the solid and liquid phases, there is therefore a need to either incorporate simultaneously some cationic species into the solid or to expel from the solid some anionic species (necessarily of phosphate nature in this case). Residual Ca
2+(aq) are expected to be rare during this dynamic washing step, especially if an excess of phosphates was used in the precipitation stage. Although the incorporation of some sodium ions cannot be ruled out, it seems logical to expect that the release of top-surface phosphate entities could be primarily involved to comply with this electroneutrality rule. Indeed, it may be assessed from
Figure 6 that both the [surface HPO
4/bulk PO
4] and the [surface PO
4/bulk PO
4] ratios decreased upon treatment. Then, Equation (5) should probably be modified into Equations (6) or (7) as follows—depending on the speciation of the released phosphate ion:
where the
species (involving H
2PO
4− ions) would arise, as considered before, from proton hopping between two adjacent HPO
42− ions due to the rather high mobility of surface protons in such nanocrystalline apatites.
In conclusion, the neutralization process appears to increase the hydroxylation of the nanocrystalline apatite while decreasing its surface acidity by removing surface protons, probably in the form of protonated phosphates to keep the electroneutrality of the system. This overall mechanistic scheme somewhat reminds the mechanism of apatite maturation proposed for nanocrystalline apatites in a previous paper [
12], and could be seen here as an accelerated transient post-maturation in alkaline conditions. Due to its strong basicity, the alkaline solution of Na
3PO
4·12H
2O pH 11 thus generates an environment propitious to neutralize the protonated acid phosphate species present within the hydrated layer to avoid acidification effects after drying and re-immersion. It may be remarked that the presence of phosphate ions in the medium upon dissolution of the Na
3PO
4·12H
2O salt may additionally contribute to reinforce this overall effect by playing a buffering tendency towards the pK
a of the HPO
42−/PO
43− acid-base couple, thus being more effective than simple washing with NaOH at pH 11 (
Figure 5).
By preventing this pH drop—potentially detrimental to cells—the addition of this (simple) equilibration step in the preparation process of (non-carbonated) biomimetic apatites thus appears as an adequate method to circumvent this acidification artefact and allow producing readily usable biomimetic apatite-based biomaterials for bone applications.
3.3. In Vitro Cytocompatibility and Antibacterial Properties via Ion Doping
In order to validate these conclusions, in vitro tests have been carried out on equilibrated samples, with the view of showing their applicability while avoiding artifacts in the (micro)biological assessments. In a first stage, we studied human osteoblast (CAL-72) cell viability. The evaluation of the non-pre-equilibrated hap-1d sample unveiled some toxicity to the cells in our working conditions (
ca. 40% cell viability, which is, thus, below the 70% limit considered as a toxicity threshold according to the ISO 10993-5:2009 standard), by evidencing a drop of the absorbance in the Neutral Red assay at t = 24 h (
Figure S3). Taking into account the biomimetic character of such non-carbonated nanocrystalline apatites, this effect may reasonably be assigned to the acidification evidenced above upon the re-immersion of dried samples. A similar test was then carried out, after pre-equilibration during the washing step with the retained methodology using Na3PO4 · 12H2O pH 11 and water. In this case, as may be seen on
Figure 7 (“non-doped” reference sample), the compound proved to be non-cytotoxic to CAL-72 osteoblasts cells, both at t = 24 and 48 h. These results clearly indicate the advantageous role of the pre-equilibration washing step, retaining the highly biocompatible compounds even in static conditions, in view of the setup of functional biomaterials.
One particularly appealing property to convey to such compounds intended as bone substitutes is antibacterial activity. This is especially important when accounting for the difficulty to eradicate established infections in bone tissue due to its highly porous character. Such antibacterial properties may be obtained by way of doping with ions such as Cu2+ or Ag+, which corresponds here to the substitute part of the Ca2+ ions by these doping ions at the time of synthesis. Antibacterial ions indeed have the advantage of avoiding bacterial resistance phenomena as opposed to antibiotics.
Copper ions are particularly appealing as they are already present in vivo (in metalloproteins) while they also exhibit other activities favoring bone regeneration such as pro-angiogenesis and osteoconduction. In this view, we have first prepared Cu
2+-doped apatite samples with increasing doping rates and post-treated them using the optimized alkaline conditions mentioned above. The main physicochemical characteristics of these doped apatite samples were investigated and are reported on
Figure S4. In particular, the “biomimetic” apatite nature of these samples was again assessed, with the physicochemical features close to those of the non-doped hap-1d and the actual doping rates found very close to the nominal rates in the precipitating solution.
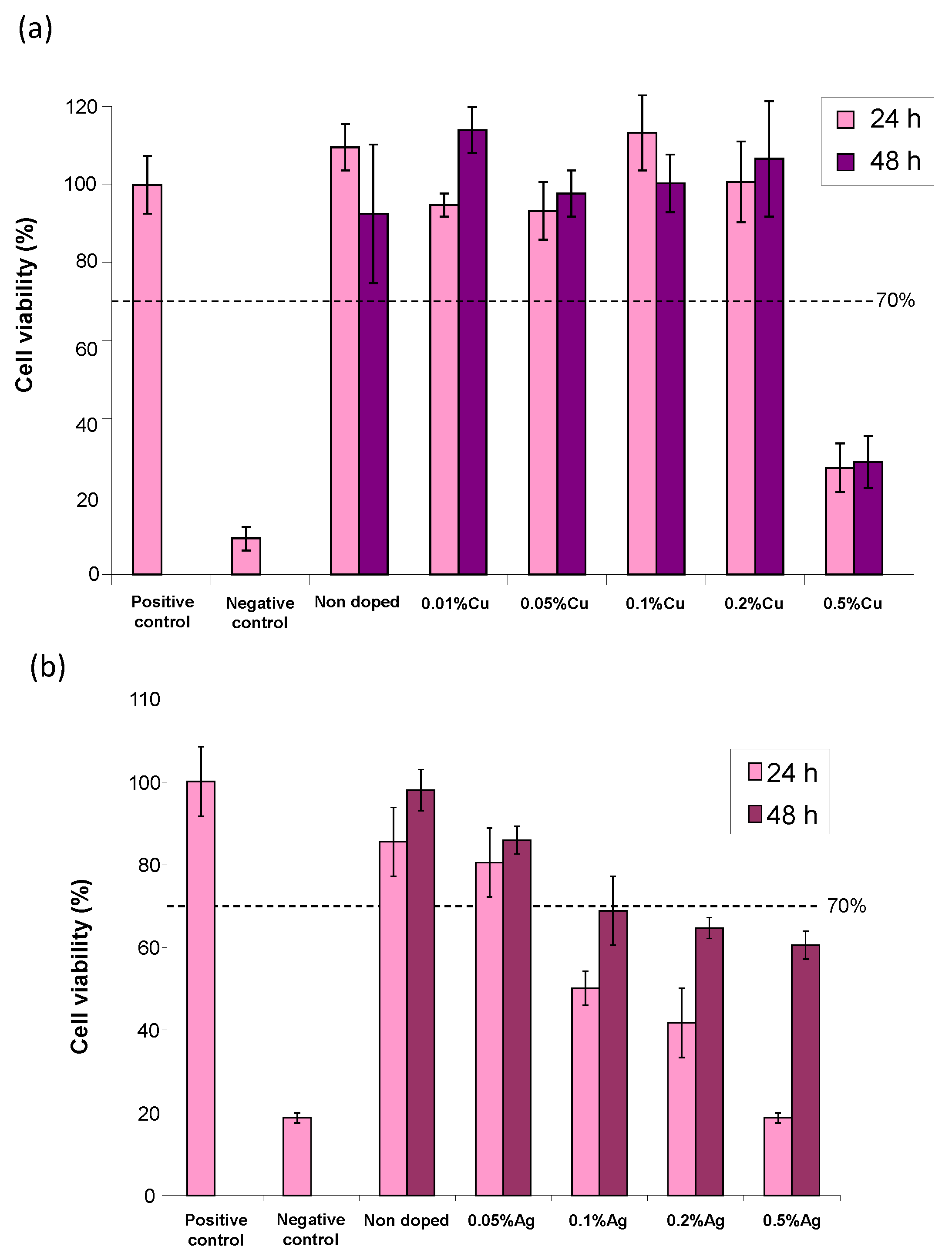
The cytocompatibility with osteoblast cells of such post-treated Cu
2+-doped apatites was then checked, first in a large range of Cu doping rates (
Figure S5a) and then in a more narrow range of interest as reported in
Figure 7a. As may be seen, except for the copper contents of 0.5 mol.% or higher, the samples with a lower Cu-doping rate led to cell viability well beyond 70% at both 24 and 48 h of contact, thus evidencing their non-cytotoxicity to CAL-72 cells.
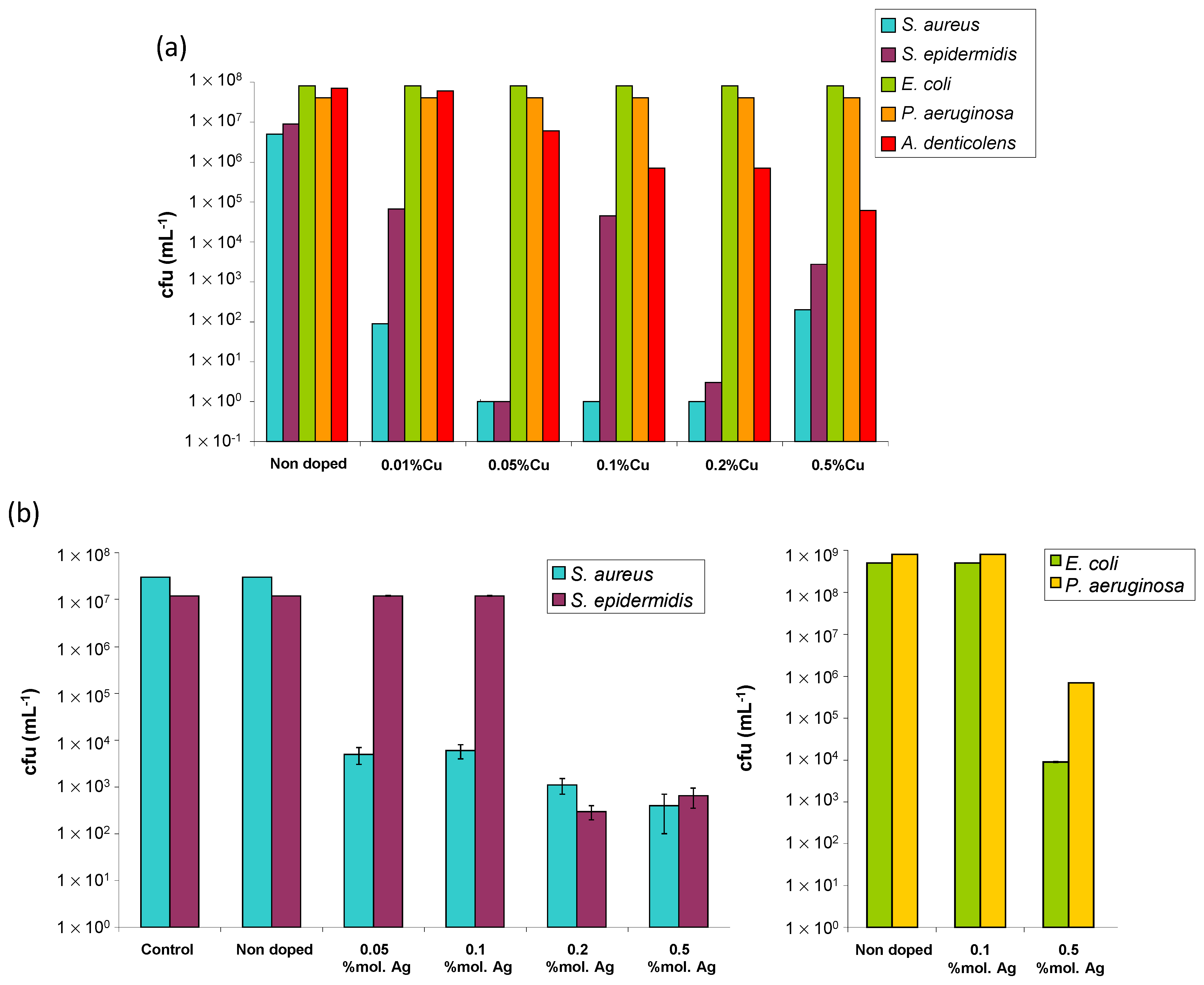
In a second step, the intrinsic antibacterial properties of these post-treated Cu-doped apatite samples were investigated on five selected bacterial strains relevant to bone infections. Four of them are aerobic bacteria, namely
S. aureus,
S. epidermidis,
E. coli, and
P. aeruginosa, and one is an aenaerobic bacterium, namely
A. denticolens (
Figure 8a). While the referenced non-doped sample did not show any intrinsic antibacterial activity, as expected, the samples doped with copper ions showed antibacterial properties with a dependence on the type of microorganism and on the dose of antibacterial agent used.
Such Cu-doped bio-inspired apatites were found to be particularly active on Gram-positive bacteria as S. aureus, S. epidermidis, and, to some extent, A. denticolens. For S. aureus, a significant decrease in bacterial colonies (exponential scale) was found for all samples, from a copper content as low as 0.01 mol.% (relative to calcium), thus leading to the elimination of more than 99.99% of the germs. For S. epidermidis, a similar trend was also found, but with a more noticeable dose-dependency. The samples with 0.01 and 0.1% of copper eliminate 99.00% of bacteria, whereas the samples with 0.5% copper eliminate 99.95% and those with copper contents between 0.05 and 0.2% eliminate more than 99.99% of the bacteria (the measure realized on the sample corresponding to 0.1% is probably artefactual). Therefore, the samples with copper contents between 0.05 and 0.2 mol.% relative to calcium are the most efficient for fighting against S. epidermidis. For A. denticolens (anaerobic), our results reveal a progressive decrease in the number of bacterial colonies as the copper content increases. If the sample with 0.01% copper appears rather inactive, the tested samples corresponding to higher copper contents (between 0.05 and 0.5% copper) lead to a decreased quantity of bacteria, with a reduction of 99.90% of the number of bacteria for a copper content of 0.5%. In contrast, for the Gram-negative E. coli and P. aeruginosa bacteria, the results obtained here did not show a significant antibacterial activity for our Cu-doped specimens as compared to the non-doped apatite.
Besides copper doping, the Ag
+-doped samples were similarly prepared and tested. The main physicochemical characteristics of these silver-bearing apatite samples were investigated (
Figure S4). As previously mentioned, the apatites close to hap-1d non-doped and exhibiting a biomimetic character were obtained. The possible incorporation of Ag
+ in apatites has been reported on several occasions [
13,
14,
15], including by some of us recently [
16]. It may, however, be remarked that the experimental doping rates found for such Ag-doped compounds were lower than the nominal ones (i.e., used in the precipitation medium). This is probably linked to the different size and charge of the Ag
+ ions compared to Ca
2+, which require structural and compositional changes in the apatitic phase [
15,
16]. In vitro assays were again conducted on CAL-72 osteoblastic cells. A progressive decreasing tendency in cell viability was observed upon increasing the Ag doping, as may be seen in
Figure S5 for a large Ag doping range. The refined results (
Figure 7b) indicate that the samples with a nominal content of Ag
+ of up to 0.5 mol.% (with reference to calcium) keep exhibiting a cell viability close to 80% at 48 h, unveiling their low toxicity. Antibacterial testing was also performed and the Ag
+-doped samples proved to significantly lower than the amount of bacterial colonies (
Figure 8b), for example, with a drop of 4 orders of magnitude for Gram-positive
S. aureus and
S. epidermidis for 0.1 and 0.2% Ag and a drop between 3 and 5 orders of magnitude for Gram-negative
E. coli and
P. aeruginosa. Thus, contrary to the Cu-doped samples studied in this work, the Ag-doped specimens were found to be both active against Gram-negative and Gram-positive bacterial strains. An SEM illustration of the anti-biofilm effect of Ag
+ doping in biomimetic apatite is for example provided in the case of the
S. aureus in
Figure 9. As may be seen, an organized biofilm is clearly visible on the non-doped sample while silver doping allows for drastically limiting the number of bacteria present on the surface and the development of an organized biofilm.
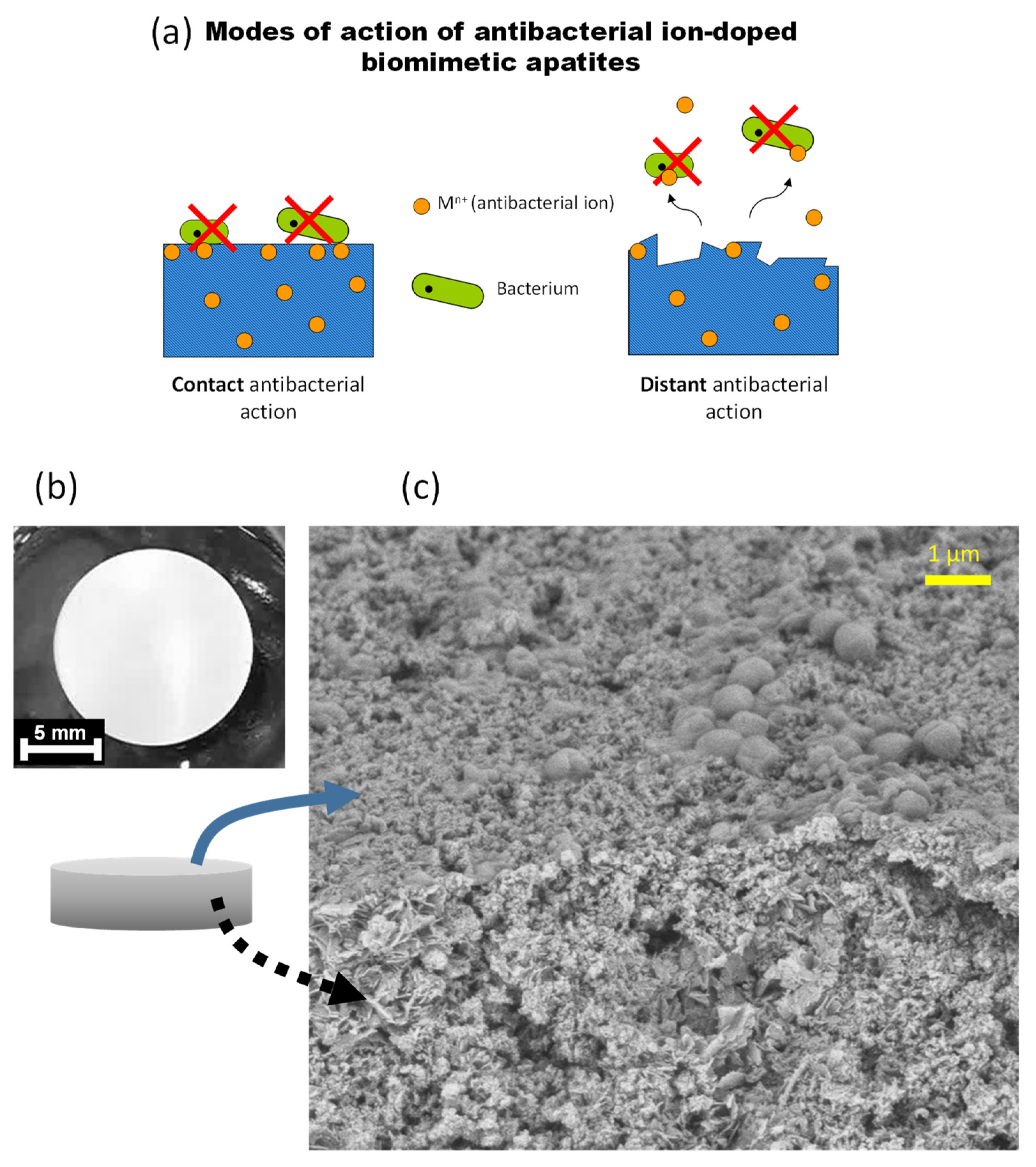
This antibacterial effect can presumably be conferred via both a “contact” mode of action (implying doping ions are exposed at the surface of the apatite crystals) and a “distant” phenomenon via the release of the antibacterial doping ions, as schematized in
Figure 10a. To explore the eventuality of this second option, the release of Ag
+ ions was followed from two Ag-doped apatite samples (hap 5% Ag matured for 20 min and 6 days) used as typical examples, in pure deionized water at RT. For both samples, about 1.2 mol.% of silver ions have been released after 6 days of immersion in simple static conditions even without cell activity, thus confirming that distant antibacterial phenomena may potentially occur. The general localization of microorganisms on typical pellets was also examined using FEG-SEM. As shown on the illustrative example provided in
Figure 10b,c, the bacteria (
S. aureus in this case) that were in contact with a hydroxyapatite pellet were found to be primarily present at the surface and not in the intercrystalline porosity. These findings further stress the relevance of a system capable of exhibiting both a “contact” and a “distant” antibacterial effect, as is the case for the ion-doped apatite samples prepared in this work.
As an intermediate summary on this part, noticeable antibacterial effects were evidenced in this work upon Cu2+ or Ag+ doping of biomimetic apatites equilibrated during the washing step with our optimized alkaline treatment. These effects could be detected (on Gram-positive strains for Cu2+ and on both Gram-positive and Gram-negative species for Ag+) at low doping rates and no “false” negative or positive data were noticed as no acidification was observed in particular for the non-doped reference sample. These findings open relevant perspectives in the field of bone repair and infection control while avoiding any acidification phenomena that are potentially detrimental to cells.



 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











