
Super-Resolution Imaging of Neuronal Structures with Structured Illumination Microscopy


Abstract
:1. Introduction
2. Methods
- cardiac perfusion with cold, freshly prepared 4% paraformaldehyde (PFA)
- fixation of the dissected brain with a 4% PFA solution on an orbital shaker overnight at 4 °C followed by washing three times with phosphate-buffered saline (PBS) at room temperature
- sectioning the brain manually using a vibratome followed by clearing of the slice with RapiClear 1.52 (SunJin Lab) overnight at room temperature
- mounting of the cleared sample with fresh RapiClear 1.52 reagent in a 0.25-mm-deep iSpacer microchamber (SunJin Lab)
3. Data Analysis
3.1. Optical Sectioning SIM (OS-SIM)
3.2. SIM with Maximum a Posteriori Probability Estimation
3.3. Spectral Merging
4. Results
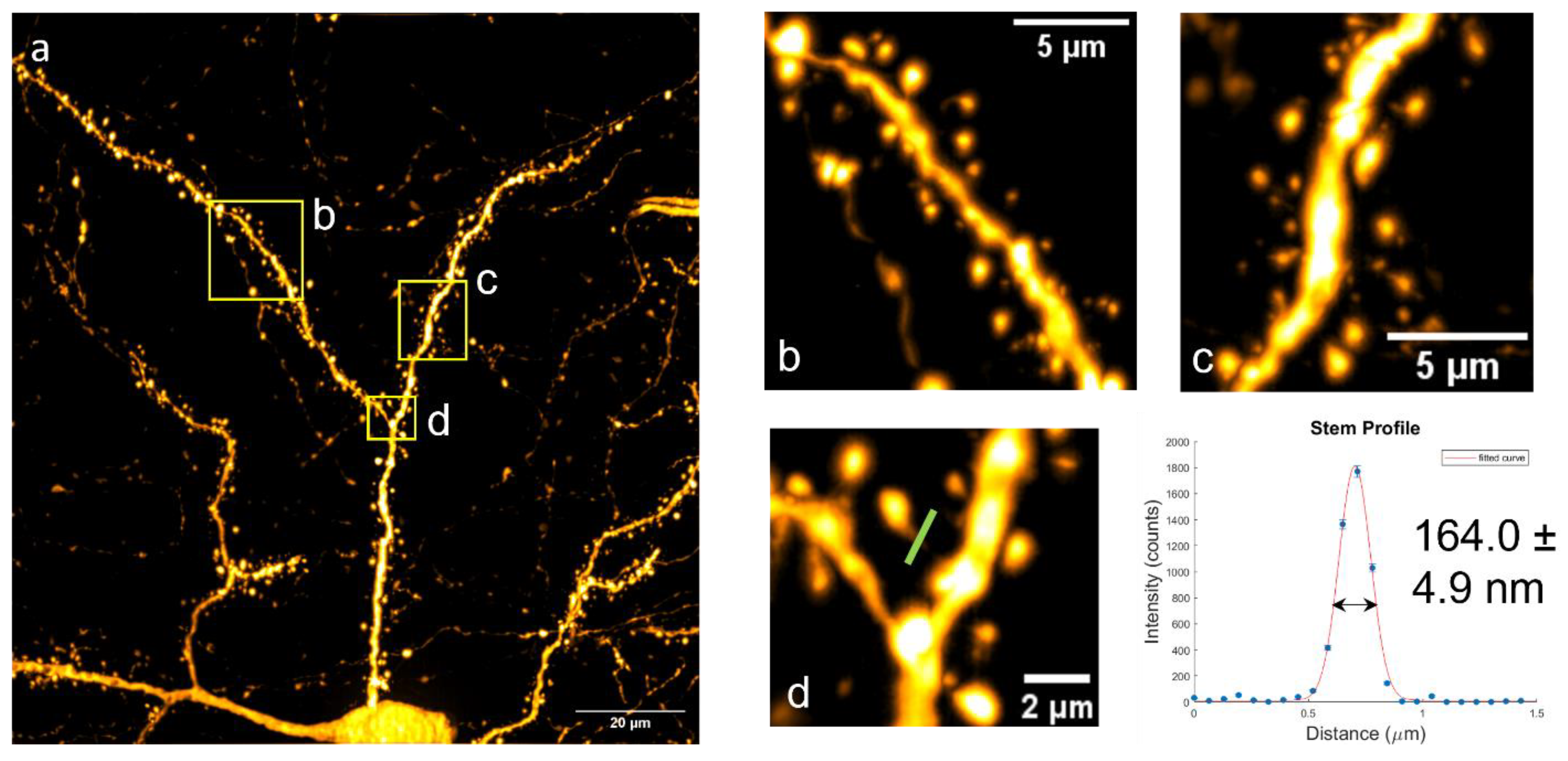
Imaging Deep Neurons
5. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Dertinger, T.; Xu, J.; Naini, O.; Vogel, R.; Weiss, S. SOFI-based 3D superresolution sectioning with a widefield microscope. Opt. Nanoscopy 2012, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.G.L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef]
- Heintzmann, R.; Cremer, C. Laterally modulated excitation microscopy: Improvement of resolution by using a diffraction grating. Proc. SPIE 1998, 3568, 185–196. [Google Scholar]
- Berning, S.; Willig, K.I.; Steffens, H.; Dibaj, P.; Hell, S.W. Nanoscopy in a Living Mouse Brain. Science 2012, 335, 551. [Google Scholar] [CrossRef]
- Mlodzianoski, M.J.; Cheng-Hathaway, P.J.; Bemiller, S.M.; McCray, T.J.; Liu, S.; Miller, D.A.; Lamb, B.T.; Landreth, G.E.; Huang, F. Active PSF shaping and adaptive optics enable volumetric localization microscopy through brain sections. Nat. Methods 2018, 15, 583–586. [Google Scholar] [CrossRef]
- Kashiwagi, Y.; Higashi, T.; Obashi, K.; Sato, Y.; Komiyama, N.H.; Grant, S.G.N.; Okabe, S. Computational geometry analysis of dendritic spines by structured illumination microscopy. Nat. Commun. 2019, 10, 1285. [Google Scholar] [CrossRef]
- Wang, M.; Tulman, D.B.; Sholl, A.B.; Kimbrell, H.Z.; Mandava, S.H.; Elfer, K.N.; Luethy, S.; Maddox, M.M.; Lai, W.; Lee, B.R.; et al. Gigapixel surface imaging of radical prostatectomy specimens for comprehensive detection of cancer-positive surgical margins using structured illumination microscopy. Sci. Rep. 2016, 6, 27419. [Google Scholar] [CrossRef]
- Gao, L.; Shao, L.; Higgins, C.D.; Poulton, J.S.; Peifer, M.; Davidson, M.W.; Wu, X.; Goldstein, B.; Betzig, E. Noninvasive imaging beyond the diffraction limit of 3D dynamics in thickly fluorescent specimens. Cell 2012, 151, 1370–1385. [Google Scholar] [CrossRef]
- Kner, P.; Chhun, B.B.; Griffis, E.R.; Winoto, L.; Gustafsson, M.G.L. Super-resolution video microscopy of live cells by structured illumination. Nat. Methods 2009, 6, 339–342. [Google Scholar] [CrossRef]
- Gustafsson, M.G.L.; Shao, L.; Carlton, P.M.; Wang, C.J.R.; Golubovskaya, I.N.; Cande, W.Z.; Agard, D.A.; Sedat, J.W. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 2008, 94, 4957–4970. [Google Scholar] [CrossRef]
- Schermelleh, L.; Carlton, P.M.; Haase, S.; Shao, L.; Winoto, L.; Kner, P.; Burke, B.; Cardoso, M.C.; Agard, D.A.; Gustafsson, M.G.L.; et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008, 320, 1332–1336. [Google Scholar] [CrossRef]
- Neil, M.A.A.; Juškaitis, R.; Wilson, T. Method of obtaining optical sectioning by using structured light in a conventional microscope. Opt. Lett. 1997, 22, 1905. [Google Scholar] [CrossRef]
- Pilger, C.; Pospíšil, J.; Müller, M.; Ruoff, M.; Schütte, M.; Spiecker, H.; Huser, T. Super-resolution fluorescence microscopy by line-scanning with an unmodified two-photon microscope. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2021, 379, 20200300. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, Q.; Zhang, Z.; Han, Y.; Kuang, C.; Xu, L.; Yang, H.; Liu, X. Three-dimensional super-resolution imaging of live whole cells using galvanometer-based structured illumination microscopy. Opt. Express 2019, 27, 7237. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.T.; Kruithoff, R.; Seedorf, G.J.; Shepherd, D.P. Multicolor structured illumination microscopy and quantitative control of polychromatic light with a digital micromirror device. Biomed. Opt. Express 2021, 12, 3700. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.J.; Meza, V.D.P.; Stelzer, E.H.K. csiLSFM combines light-sheet fluorescence microscopy and coherent Structured illumination for a lateral resolution below 100 nm. Proc. Natl. Acad. Sci. USA 2017, 114, 4869–4874. [Google Scholar] [CrossRef] [PubMed]
- Křížek, P.; Raška, I.; Hagen, G.M. Flexible structured illumination microscope with a programmable illumination array. Opt. Express 2012, 20, 24585. [Google Scholar] [CrossRef] [PubMed]
- Rossberger, S.; Best, G.; Baddeley, D.; Heintzmann, R.; Birk, U.; Dithmar, S.; Cremer, C. Combination of structured illumination and single molecule localization microscopy in one setup. J. Opt. 2013, 15, 094003. [Google Scholar] [CrossRef]
- Young, L.J.; Ströhl, F.; Kaminski, C.F. A Guide to Structured Illumination TIRF Microscopy at High Speed with Multiple Colors. J. Vis. Exp. 2016, 111, e53988. [Google Scholar]
- Poher, V.; Zhang, H.X.; Kennedy, G.T.; Griffin, C.; Oddos, S.; Gu, E.; Elson, D.S.; Girkin, M.; French, P.M.W.; Dawson, M.D.; et al. Optical sectioning microscope with no moving parts using a micro-stripe array light emitting diode. Opt. Express 2007, 15, 11196–11206. [Google Scholar] [CrossRef] [PubMed]
- Pospíšil, J.; Wiebusch, G.; Fliegel, K.; Klíma, M.; Huser, T. Highly compact and cost-effective 2-beam super-resolution structured illumination microscope based on all-fiber optic components. Opt. Express 2021, 29, 11833. [Google Scholar] [CrossRef]
- Hinsdale, T.A.; Stallinga, S.; Rieger, B. High-speed multicolor structured illumination microscopy using a hexagonal single mode fiber array. Biomed. Opt. Express 2021, 12, 1181. [Google Scholar] [CrossRef]
- Lukeš, T.; Křížek, P.; Švindrych, Z.; Benda, J.; Ovesný, M.; Fliegel, K.; Klíma, M.; Hagen, G.M. Three-dimensional super-resolution structured illumination microscopy with maximum a posteriori probability image estimation. Opt. Express 2014, 22, 29805–29817. [Google Scholar] [CrossRef]
- Orieux, F.; Sepulveda, E.; Loriette, V.; Dubertret, B.; Olivo-Marin, J.C. Bayesian estimation for optimized structured illumination microscopy. IEEE Trans. Image Process. 2012, 21, 601–614. [Google Scholar] [CrossRef]
- Huang, X.; Fan, J.; Li, L.; Liu, H.; Wu, R.; Wu, Y.; Wei, L.; Mao, H.; Lal, A.; Xi, P.; et al. Fast, long-term, super-resolution imaging with Hessian structured illumination microscopy. Nat. Biotechnol. 2018, 36, 451–459. [Google Scholar] [CrossRef]
- Lukeš, T.; Hagen, G.M.; Křížek, P.; Švindrych, Z.; Fliegel, K.; Klíma, M. Comparison of image reconstruction methods for structured illumination microscopy. Proc. SPIE 2014, 9129, 91293J. [Google Scholar]
- Chakrova, N.; Rieger, B.; Stallinga, S. Deconvolution methods for structured illumination microscopy. J. Opt. Soc. Am. A 2016, 33, B12. [Google Scholar] [CrossRef]
- Chen, X.; Zhong, S.; Hou, Y.; Cao, R.; Wang, W.; Li, D.; Dai, Q.; Kim, D.; Xi, P. Superresolution structured illumination microscopy reconstruction algorithms: A review. Light Sci. Appl. 2023, 12, 172. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Wang, K.; Li, L.; Xing, S.; Ye, S.; Wen, J.; Duan, X.; Luo, Z.; Gou, W.; Chen, T.; et al. Quantitative structured illumination microscopy via a physical model-based background filtering algorithm reveals actin dynamics. Nat. Commun. 2023, 14, 3089. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Li, Y.; Chen, X.; Ge, X.; Li, M.; Guan, M.; Hou, Y.; Fu, Y.; Xu, X.; Jiang, S.; et al. Open-3DSIM: An open-source three-dimensional structured illumination microscopy reconstruction platform. Nat. Methods 2023, 20, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Hannebelle, M.T.; Raeth, E.; Leitao, S.M.; Lukeš, T.; Pospíšil, J.; Toniolo, C.; Venzin, O.F.; Chrisnandy, A.; Swain, P.P.; Ronceray, N.; et al. OpenSIM: Open source microscope add-on for structured illumination microscopy. bioRxiv 2023, arXiv:2023.06.16.545316. [Google Scholar]
- Wen, G.; Li, S.; Liang, Y.; Wang, L.; Zhang, J.; Chen, X.; Jin, X.; Chen, C.; Tang, Y.; Li, H. Spectrum-optimized direct image reconstruction of super-resolution structured illumination microscopy. PhotoniX 2023, 4, 19. [Google Scholar] [CrossRef]
- Li, X.; Wu, Y.; Su, Y.; Rey-Suarez, I.; Matthaeus, C.; Updegrove, T.B.; Wei, Z.; Zhang, L.; Sasaki, H.; Li, Y.; et al. Three-dimensional structured illumination microscopy with enhanced axial resolution. Nat. Biotechnol. 2023, 2023, 1–13. [Google Scholar] [CrossRef]
- Johnson, K.A.; Noble, D.; Machado, R.; Paul, T.C.; Hagen, G.M. Flexible Multiplane Structured Illumination Microscope with a Four-Camera Detector. Photonics 2022, 9, 501. [Google Scholar] [CrossRef]
- Luo, F.; Zeng, J.; Shao, Z.; Zhang, C. Fast structured illumination microscopy via transfer learning with correcting. Opt. Lasers Eng. 2023, 162, 107432. [Google Scholar] [CrossRef]
- Burns, Z.; Liu, Z.; Liu, Z.; Liu, Z.; Liu, Z. Untrained, physics-informed neural networks for structured illumination microscopy. Opt. Express 2023, 31, 8714–8724. [Google Scholar] [CrossRef]
- Pospíšil, J.; Lukeš, T.; Bendesky, J.; Fliegel, K.; Spendier, K.; Hagen, G.M. Imaging tissues and cells beyond the diffraction limit with structured illumination microscopy and Bayesian image reconstruction. Gigascience 2018, 8, giy126. [Google Scholar] [CrossRef]
- Wu, Y.; Shroff, H. Faster, sharper, and deeper: Structured illumination microscopy for biological imaging. Nat. Methods 2018, 15, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Mandula, O.; Kielhorn, M.; Wicker, K.; Krampert, G.; Kleppe, I.; Heintzmann, R. Line scan—Structured illumination microscopy super-resolution imaging in thick fluorescent samples. Opt. Express 2012, 20, 24167–24174. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Q.; Chou, S.W.; Newman, Z.; Turcotte, R.; Natan, R.; Dai, Q.; Isacoff, E.Y.; Ji, N. Fast widefield imaging of neuronal structure and function with optical sectioning in vivo. Sci. Adv. 2020, 6, eaaz3870. [Google Scholar] [CrossRef]
- Lu, R.; Liang, Y.; Meng, G.; Zhou, P.; Svoboda, K.; Paninski, L.; Ji, N. Rapid mesoscale volumetric imaging of neural activity with synaptic resolution. Nat. Methods 2020, 17, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Křížek, P.; Lukeš, T.; Ovesný, M.; Fliegel, K.; Hagen, G.M. SIMToolbox: A MATLAB toolbox for structured illumination fluorescence microscopy. Bioinformatics 2015, 32, 318–320. [Google Scholar] [CrossRef]
- Johnson, K.A.; Hagen, G.M. Artifact-free whole-slide imaging with structured illumination microscopy and Bayesian image reconstruction. Gigascience 2021, 9, giaa035. [Google Scholar] [CrossRef]
- Heintzmann, R. Structured illumination methods. In Handbook of Biological Confocal Microscopy; Pawley, J.B., Ed.; Springer: New York, NY, USA, 2006; pp. 265–279. [Google Scholar]
- Šonka, M.; Hlaváč, V.; Boyle, R. Image Processing Analysis and Machine Vision, 2nd ed.; PWS Publishing: Boston, MA, USA, 1998. [Google Scholar]
- Goodman, J.W. Frequency Analysis of Optical Imaging Systems. In Introduction to Fourier Optics; McGraw-HIll Int.: New York, NY, USA, 1968; pp. 126–171. ISBN 0-07-024254-2. [Google Scholar]
- Pospíšil, J.; Fliegel, K.; Klíma, M. Assessing resolution in live cell structured illumination microscopy. In Proceedings of SPIE—The International Society for Optical Engineering; Páta, P., Fliegel, K., Eds.; SPIE: Prague, Czech Republic, 2017; Volume 10603, p. 39. [Google Scholar]
- Verveer, P.J.; Jovin, T.M. Efficient superresolution restoration algorithms using maximum a posteriori estimations with application to fluorescence microscopy. JOSA A 1997, 14, 1696. [Google Scholar] [CrossRef]
- Verveer, P.J.; Gemkow, M.J.; Jovin, T.M. A comparison of image restoration approaches applied to three-dimensional confocal and wide-field fluorescence microscopy. J. Microsc. 1999, 193, 50–61. [Google Scholar] [CrossRef]
- Vermolen, B.J.; Garini, Y.; Young, I.T. 3D restoration with multiple images acquired by a modified conventional microscope. Microsc. Res. Tech. 2004, 64, 113–125. [Google Scholar] [CrossRef]
- Chaudhuri, S. Super-Resolution Imaging; Milanfar, P., Ed.; CRC Press: Boca Raton, FL, USA, 2011; ISBN 978-1-4398-1931-9. [Google Scholar]
- Preibisch, S.; Saalfeld, S.; Tomancak, P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 2009, 25, 1463–1465. [Google Scholar] [CrossRef]
- Geissbuehler, M.; Lasser, T. How to display data by color schemes compatible with red-green color perception deficiencies. Opt. Express 2013, 21, 9862. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Franklin, K.B.J. The Mouse Brain in Stereotaxic Coordinates, 2nd ed.; Academic Press: New York, NY, USA, 2001; ISBN 9780128161579. [Google Scholar]
- Allen Institute for Brain Science (2004). Allen Institute for Brain Science (2011). Allen Mouse Brain Atlas [Dataset]. Available online: https://mouse.brain-map.org/ (accessed on 24 May 2023).
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef]
- Nieuwenhuizen, R.P.J.; Lidke, K.A.; Bates, M.; Puig, D.L.; Grünwald, D.; Stallinga, S.; Rieger, B. Measuring image resolution in optical nanoscopy. Nat. Methods 2013, 10, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Culley, S.; Albrecht, D.; Jacobs, C.; Pereira, P.M.; Leterrier, C.; Mercer, J.; Henriques, R. Quantitative mapping and minimization of super-resolution optical imaging artifacts. Nat. Methods 2018, 15, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, E.; Borkovec, J.; Kováčik, L.; Svidenská, S.; Schröfel, A.; Skalníková, M.; Švindrych, Z.; Křížek, P.; Ovesný, M.; Hagen, G.M.; et al. Separation of replication and transcription domains in nucleoli. J. Struct. Biol. 2014, 188, 259–266. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNR (dB) | Resolution (nm) | |
|---|---|---|
| Widefield | 43.87 | 247.6 |
| Basic SIM | 29.33 | 251.6 |
| MAP-SIM | 39.27 | 143.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paul, T.C.; Johnson, K.A.; Hagen, G.M. Super-Resolution Imaging of Neuronal Structures with Structured Illumination Microscopy. Bioengineering 2023, 10, 1081. https://doi.org/10.3390/bioengineering10091081
Paul TC, Johnson KA, Hagen GM. Super-Resolution Imaging of Neuronal Structures with Structured Illumination Microscopy. Bioengineering. 2023; 10(9):1081. https://doi.org/10.3390/bioengineering10091081
Chicago/Turabian StylePaul, Tristan C., Karl A. Johnson, and Guy M. Hagen. 2023. "Super-Resolution Imaging of Neuronal Structures with Structured Illumination Microscopy" Bioengineering 10, no. 9: 1081. https://doi.org/10.3390/bioengineering10091081
APA StylePaul, T. C., Johnson, K. A., & Hagen, G. M. (2023). Super-Resolution Imaging of Neuronal Structures with Structured Illumination Microscopy. Bioengineering, 10(9), 1081. https://doi.org/10.3390/bioengineering10091081