
ResnetAge: A Resnet-Based DNA Methylation Age Prediction Method


Abstract
:1. Introduction
2. Materials and Methods
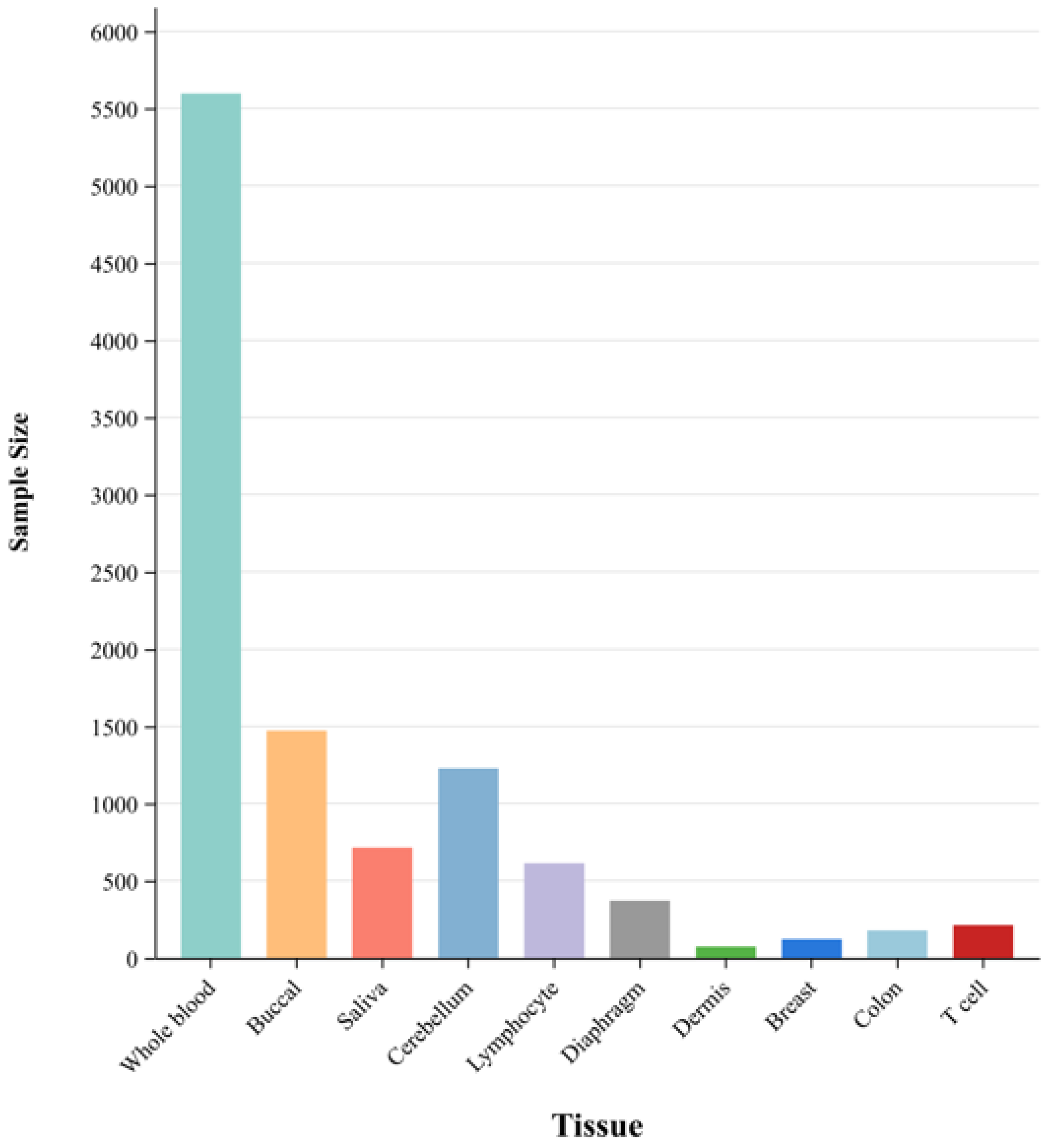
2.1. Data Source and CpG Site Selection
2.2. Data Processing
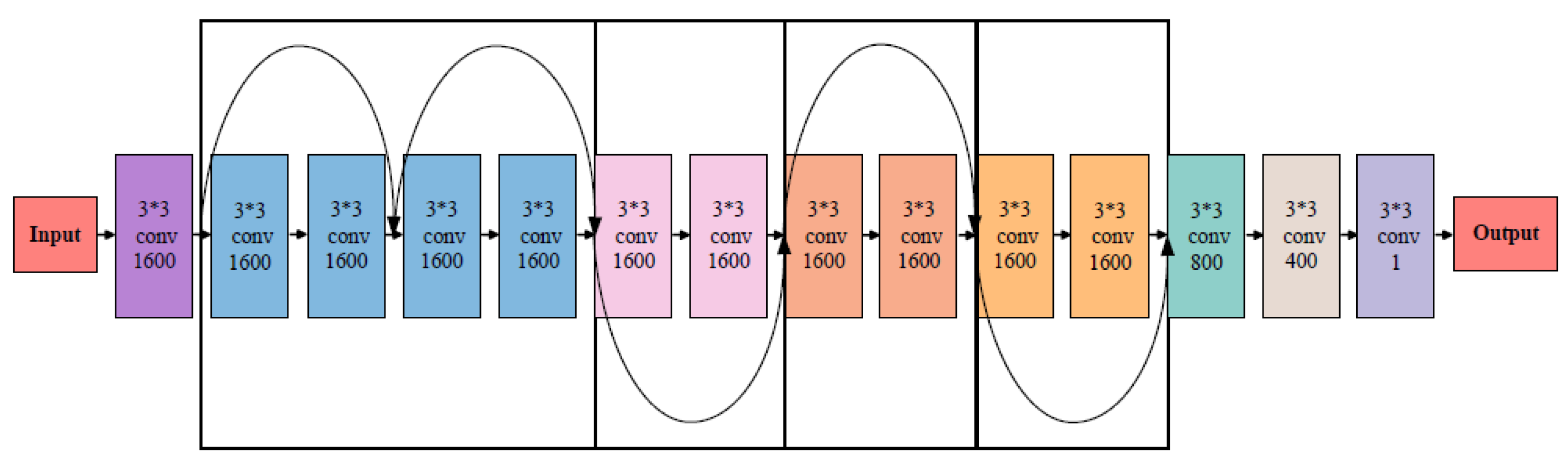
2.3. Model Design
2.4. Model Training Process
3. Results Analysis
3.1. Evaluation Indicators
3.2. Model Training Results
3.3. Comparative Results of Different Methods
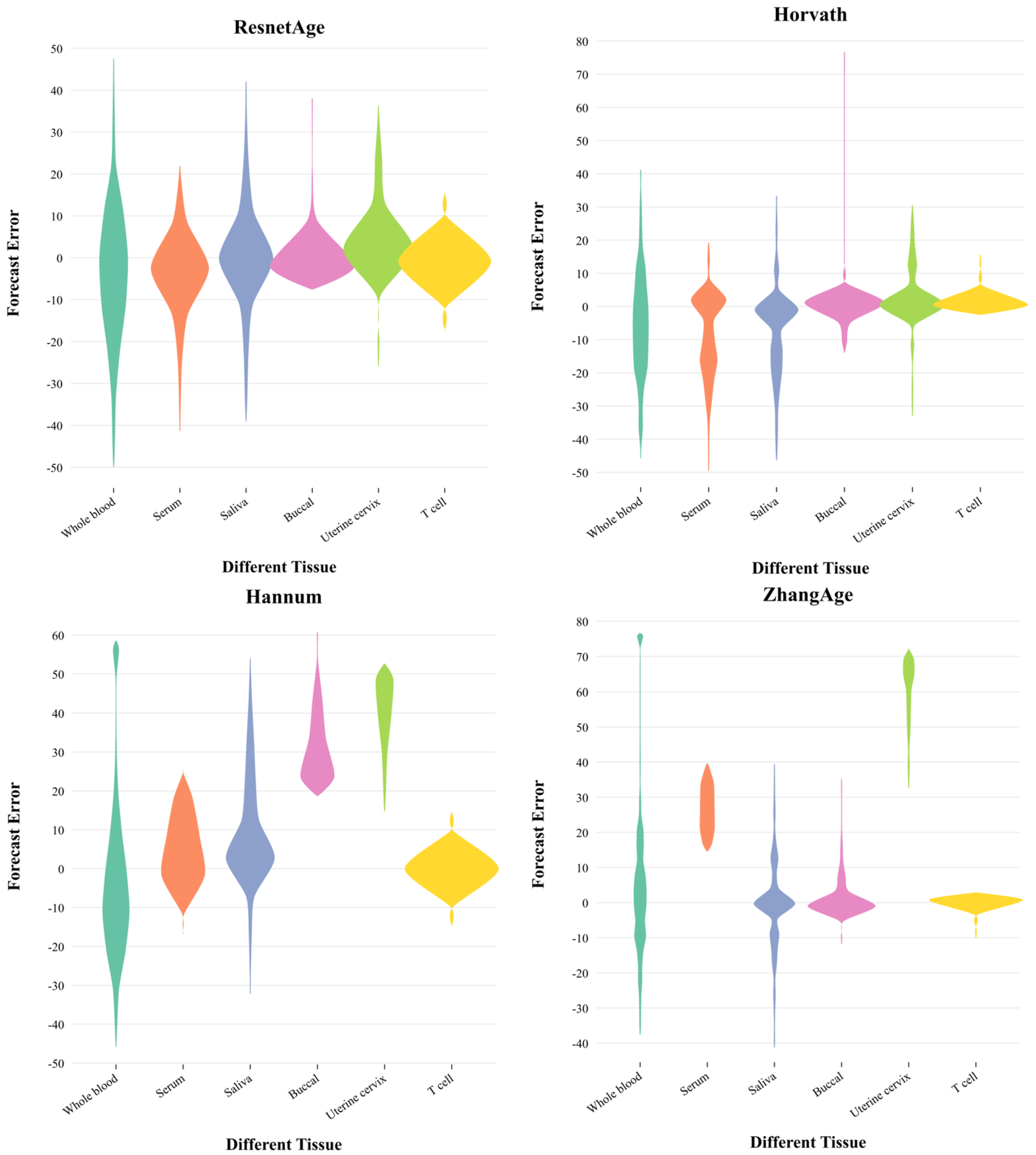
3.4. Prediction Performance in Different Tissues
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- De Lima, C.L.P.; Lapierre, L.R.; Singh, R. A pan-tissue DNA-methylation epigenetic clock based on deep learning. NPJ Aging 2022, 8, 4. [Google Scholar] [CrossRef]
- Wei, Z.; Ding, S.; Duan, M.; Liu, S.; Huang, L.; Zhou, F. FeSTwo, a two-step feature selection algorithm based on feature engineering and sampling for the chronological age regression problem. Comput. Biol. Med. 2020, 125, 104008. [Google Scholar] [CrossRef] [PubMed]
- Benayoun, B.A.; Pollina, E.A.; Brunet, A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef] [PubMed]
- Jylhävä, J.; Pedersen, N.; Hägg, S. Biological age predictors. EBioMedicine 2017, 21, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Ding, S.; Duan, M.; Liu, S.; Huang, L.; Zhou, F. Longitudinal trajectories, correlations and mortality associations of nine biological ages across 20-years follow-up. Elife 2020, 9, e51507. [Google Scholar]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, 3156. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, T.M.; Bonder, M.J.; Stark, A.K.; Krueger, F.; von Meyenn, F.; Stegle, O.; Reik, W. Multi-tissue DNA methylation age predictor in mouse. Genome Biol. 2017, 18, 68. [Google Scholar] [CrossRef]
- Thompson, M.J.; Chwiałkowska, K.; Rubbi, L.; Lusis, A.J.; Davis, R.C.; Srivastava, A.; Korstanje, R.; Churchill, G.A.; Horvath, S.; Pellegrini, M. A multi-tissue full lifespan epigenetic clock for mice. Aging 2018, 10, 2832. [Google Scholar] [CrossRef]
- Thompson, M.J.; Horvath, S.; Pellegrini, M. An epigenetic aging clock for dogs and wolves. Aging 2017, 9, 1055. [Google Scholar] [CrossRef]
- Debrabant, B.; Soerensen, M.; Christiansen, L.; Tan, Q.; McGue, M.; Christensen, K.; Hjelmborg, J. DNA methylation age and perceived age in elderly Danish twins. Mech. Ageing Dev. 2018, 169, 40–44. [Google Scholar] [CrossRef]
- Beach, S.R.; Dogan, M.V.; Lei, M.K.; Cutrona, C.E.; Gerrard, M.; Gibbons, F.X.; Simons, R.L.; Brody, G.H.; Philibert, R.A. Methylomic aging as a window onto the influence of lifestyle: Tobacco and alcohol use alter the rate of biological aging. J. Am. Geriatr. Soc. 2015, 63, 2519–2525. [Google Scholar] [CrossRef] [PubMed]
- Fakouri, N.B.; Hou, Y.; Demarest, T.G.; Christiansen, L.S.; Okur, M.N.; Mohanty, J.G.; Croteau, D.L.; Bohr, V.A. Toward understanding genomic instability, mitochondrial dysfunction and aging. FEBS J. 2019, 286, 1058–1073. [Google Scholar] [CrossRef] [PubMed]
- Ximerakis, M.; Lipnick, S.L.; Innes, B.T.; Simmons, S.K.; Adiconis, X.; Dionne, D.; Mayweather, B.A.; Nguyen, L.; Niziolek, Z.; Ozek, C.; et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci. 2019, 22, 1696–1708. [Google Scholar] [CrossRef] [PubMed]
- Alisch, R.S.; Barwick, B.G.; Chopra, P.; Myrick, L.K.; Satten, G.A.; Conneely, K.N.; Warren, S.T. Age-associated DNA methylation in pediatric populations. Genome Res. 2012, 22, 623–632. [Google Scholar] [CrossRef]
- Bockl, t.S.; Lin, W.; Sehl, M.E.; Sánchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic predictor of age. PLoS ONE 2011, 6, e14821. [Google Scholar]
- Bollati, V.; Schwartz, J.; Wright, R.; Litonjua, A.; Tarantini, L.; Suh, H.; Sparrow, D.; Vokonas, P.; Baccarelli, A. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech. Ageing Dev. 2009, 130, 234–239. [Google Scholar] [CrossRef]
- Jung, S.E.; Shin, K.J.; Lee, H.Y. DNA methylation-based age prediction from various tissues and body fluids. BMB Rep. 2017, 50, 546. [Google Scholar] [CrossRef]
- Vidaki, A.; Ballard, D.; Aliferi, A.; Miller, T.H.; Barron, L.P.; Court, D.S. DNA methylation-based forensic age prediction using artificial neural networks and next generation sequencing. Forensic Sci. Int. Genet. 2017, 28, 225–236. [Google Scholar] [CrossRef]
- Bork, S.; Pfister, S.; Witt, H.; Horn, P.; Korn, B.; Ho, A.D.; Wagner, W. DNA methylation pattern changes upon long-term culture and aging of human mesenchymal stromal cells. Aging Cell 2010, 9, 54–63. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.I.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Vallerga, C.L.; Walker, R.M.; Lin, T.; Henders, A.K.; Montgomery, G.W.; He, J.; Fan, D.; Fowdar, J.; Kennedy, M.; et al. Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Med. 2019, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B Stat. Methodol. 2005, 67, 301–320. [Google Scholar] [CrossRef]
- Holzscheck, N.; Falckenhayn, C.; Söhle, J.; Kristof, B.; Siegner, R.; Werner, A.; Schössow, J.; Jürgens, C.; Völzke, H.; Wenck, H.; et al. Modeling transcriptomic age using knowledge-primed artificial neural networks. NPJ Aging Mech. Dis. 2021, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Putin, E.; Mamoshina, P.; Aliper, A.; Korzinkin, M.; Moskalev, A.; Kolosov, A.; Ostrovskiy, A.; Cantor, C.; Vijg, J.; Zhavoronkov, A. Deep biomarkers of human aging: Application of deep neural networks to biomarker development. Aging 2016, 8, 1021. [Google Scholar] [CrossRef]
- Galkin, F.; Mamoshina, P.; Kochetov, K.; Sidorenko, D.; Zhavoronkov, A. DeepMAge: A methylation aging clock developed with deep learning. Aging Dis. 2021, 12, 1252. [Google Scholar] [CrossRef]
- Levy, J.J.; Titus, A.J.; Petersen, C.L.; Chen, Y.; Salas, L.A.; Christensen, B.C. MethylNet: An automated and modular deep learning approach for DNA methylation analysis. BMC Bioinform. 2020, 21, 108. [Google Scholar] [CrossRef]
- Di Lena, P.; Sala, C.; Prodi, A.; Nardini, C. Missing value estimation methods for DNA methylation data. Bioinformatics 2019, 35, 3786–3793. [Google Scholar] [CrossRef]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 26 June–1 July 2016; pp. 770–778. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Platform | MAE ResnetAge | MAD ResnetAge | MAE Horvath | MAD Horvath | MAE Hannum | MAD Hannum | MAE ZhangAge | MAD ZhangAge |
|---|---|---|---|---|---|---|---|---|---|
| GSE57484 | 27K | 6.93 | 6.62 | 8.63 | 8.88 | \ | \ | \ | \ |
| GSE58119 | 27K | 8.39 | 6.77 | 13.99 | 12.84 | \ | \ | \ | \ |
| GSE137495 | 450K | 0.26 | 0.25 | 0.59 | 0.56 | 26.22 | 26.2 | 1.31 | 0.94 |
| GSE80261 | 450K | 4.39 | 3.34 | 3.84 | 3.14 | 36.24 | 35.93 | 4.74 | 4.48 |
| GSE111223 | 450K | 10.48 | 8.48 | 10.54 | 9.73 | 12.08 | 11.07 | 0.9 | 0.78 |
| GSE71245 | 450K | 8.17 | 5.44 | 7.17 | 5.67 | 10.27 | 9.29 | 5.44 | 6.14 |
| GSE53740 | 450K | 12.02 | 9.9 | 8.22 | 7.28 | 8.82 | 8.43 | 0.48 | 0.49 |
| GSE30758 | 27K | 11.89 | 10.9 | 7.2 | 7.11 | \ | \ | \ | \ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, L.; Hai, B.; Kuang, Z.; Wang, H.; Zhao, J. ResnetAge: A Resnet-Based DNA Methylation Age Prediction Method. Bioengineering 2024, 11, 34. https://doi.org/10.3390/bioengineering11010034
Shi L, Hai B, Kuang Z, Wang H, Zhao J. ResnetAge: A Resnet-Based DNA Methylation Age Prediction Method. Bioengineering. 2024; 11(1):34. https://doi.org/10.3390/bioengineering11010034
Chicago/Turabian StyleShi, Lijuan, Boquan Hai, Zhejun Kuang, Han Wang, and Jian Zhao. 2024. "ResnetAge: A Resnet-Based DNA Methylation Age Prediction Method" Bioengineering 11, no. 1: 34. https://doi.org/10.3390/bioengineering11010034
APA StyleShi, L., Hai, B., Kuang, Z., Wang, H., & Zhao, J. (2024). ResnetAge: A Resnet-Based DNA Methylation Age Prediction Method. Bioengineering, 11(1), 34. https://doi.org/10.3390/bioengineering11010034