1. Introduction
Antibodies have made a revolutionary impact in the realms of science and medicine because of their remarkable specificity and biochemical adaptability, making them applicable across a wide spectrum of biomedical investigations. The conventional antibody structure comprises two heavy chains and two light chains, each housing a variable domain denoted as VH and VL, respectively. A notable deviation from this typical architecture is observed in camelids (llamas, camels, alpacas, and their kins), which possess an antibody repertoire exclusively composed of heavy chains [
1]. In this unique system, antibodies bind to their target antigens using a single variable domain of heavy-chain-only antibody (VHH, also known as a nanobody). A nanobody has a smaller size (∼10–15 kDa) compared to the traditional fragment of antigen binding (Fab) (∼50 kDa). In addition, it does not rely on heavy–light chain pairing, which renders it highly suitable for constructing polyspecific antibodies from a protein engineering perspective. Unlike the Fab of conventional antibodies, a nanobody can be efficiently expressed in bacteria. The versatility of nanobodies extends their utility to various applications in protein structural biology and cell biology, positioning them as potential diagnostic and therapeutic agents [
2].
Despite the increasing significance of nanobodies in biomedical research, existing methods for producing monoclonal nanobody sequences involve the immunization of camelids, which remains tedious, costly, and frequently unavailable. Additionally, antibodies obtained from immunization often face limitations in binding immunodominant epitopes. The identification of functional clones, such as conformationally selective nanobodies, remains a challenging task [
3,
4]. In response to these challenges, synthetic libraries have been designed for the in vitro selection against diverse antigens [
4,
5,
6,
7]. In this study, we also devised a synthetic nanobody library in a phage display format for the in vitro selection of antigen-specific nanobodies. Unlike most of the other synthetic nanobody libraries, selected complementarity determining region (CDR) positions in our library were randomized using degenerate codon. This design performs complete randomization of these sites, which may allow for the selection of CDR sequences not captured by a pre-biased library with a planned diversity introduced by a combination of trinucleotide phosphoramides [
3,
4,
5,
6,
8,
9].
To validate the efficacy of our proposed libraries, we executed antibody discovery against an anti-tumor drug target: CD276 (also denoted as B7-H3). It is a member of the B7 ligand family, emerging as a promising target for antibody-based immunotherapy [
10,
11]. The prevailing variant of human B7-H3 features four immunoglobulin-like domains (Ig) in a tandemly repeated format such as IgV-IgC-IgV-IgC, while human B7-H3 exists in two isoforms (2IgB7-H3 and 4IgB7-H3); its mouse counterpart has a singular 2Ig isoform [
12]. B7-H3 is prominently expressed in both differentiated malignant cells and cancer-initiating cells, exhibiting limited heterogeneity but a high frequency across various cancer types [
11]. Importantly, its expression in normal tissues is low. In nonmalignant tissues, B7-H3 primarily assumes an inhibitory role in adaptive immunity by suppressing T-cell activation and proliferation [
13,
14]. In malignant tissues, B7-H3 hampers tumor antigen-specific immune responses, fostering a tumor-promoting effect [
11]. Beyond its immunologic functions, B7-H3 also facilitates migration and invasion [
15], promoting angiogenesis [
16,
17], enhancing chemoresistance [
18,
19], influencing tumor cell metabolism [
20,
21], and triggering endothelial-to-mesenchymal transition [
22,
23]. Consequently, the presence of B7-H3 in tumors correlates with a poor prognosis [
17,
24]. Despite experimental evidence indicating that silencing B7-H3 reduces the malignant potential of cancer cells, the development of B7-H3-blocking antibodies has received limited attention, largely due to the unknown B7-H3 receptor. Instead, numerous antibody-based strategies have been devised leveraging distinct effector mechanisms against B7-H3-expressing cancer cells [
11].
Antibodies targeting distinct epitopes can show different modes of actions and affect potencies [
25,
26]. Both the mouse-derived anti-human B7-H3 monoclonal antibodies (MAbs) 8H9 and 376.96, which underwent clinical testing, were identified as binding to the FG loop region at the top of the IgV domain [
27,
28]. The single-chain structure of nanobodies contributes to the convex shape of the paratopes. In comparison, the paratopes of conventional antibodies are generally wider and form a flat surface, a groove or cavity due to the heavy and light chain pairing. Thus, the footprint of the paratope of a nanobody on its cognate antigen might be different from that of conventional antibodies. In this study, we took advantage of the synthetic nanobody library, aiming to discover B7-H3-specific nanobodies which might bind to different epitopes compared to known MAbs. The synthetic phage display nanobody library was first evaluated by next-generation sequencing (NGS) to ensure diversity. A phage display selection was then conducted to enrich binders for recombinant human B7-H3 proteins. Both single-clone phage enzyme-linked immunosorbent assay (ELISA) and NGS were applied to identify binders. Subsequently, the positive clones were characterized for their affinity, specificity, and potential binding sites. Lastly, one of the nanobodies with a higher binding affinity was converted to a human IgG1 fragment crystallizable (Fc) domain-fusion antibody and tested in an antibody-dependent cellular cytotoxicity (ADCC) reporter assay to demonstrate its functional activity. As outlined in this report, the described library exhibits high productivity and holds the potential to generate specific MAbs targeting distinct epitopes.
3. Discussion
Various reasons support the utilization of nanobodies in monoclonal antibody, bispecific, or chimeric antigen receptor (CAR) T cell therapy developments, such as their distinctive single-chain structure, compact size, and ease of expression and production [
29]. In this study, we successfully isolated nanobodies from our synthetic phage display library, which used degenerate codon for CDR randomization. One previous study also used degenerate codon for nanobody CDR randomization but in a ribosome display library with a higher library size (
) [
7]. Ribosome display is not limited by cell transformation and culture constraints, allowing for a much higher diversity for display, but it remains underutilized compared to phage display or yeast display systems, possibly due to its sub-optimal efficiency and fidelity.
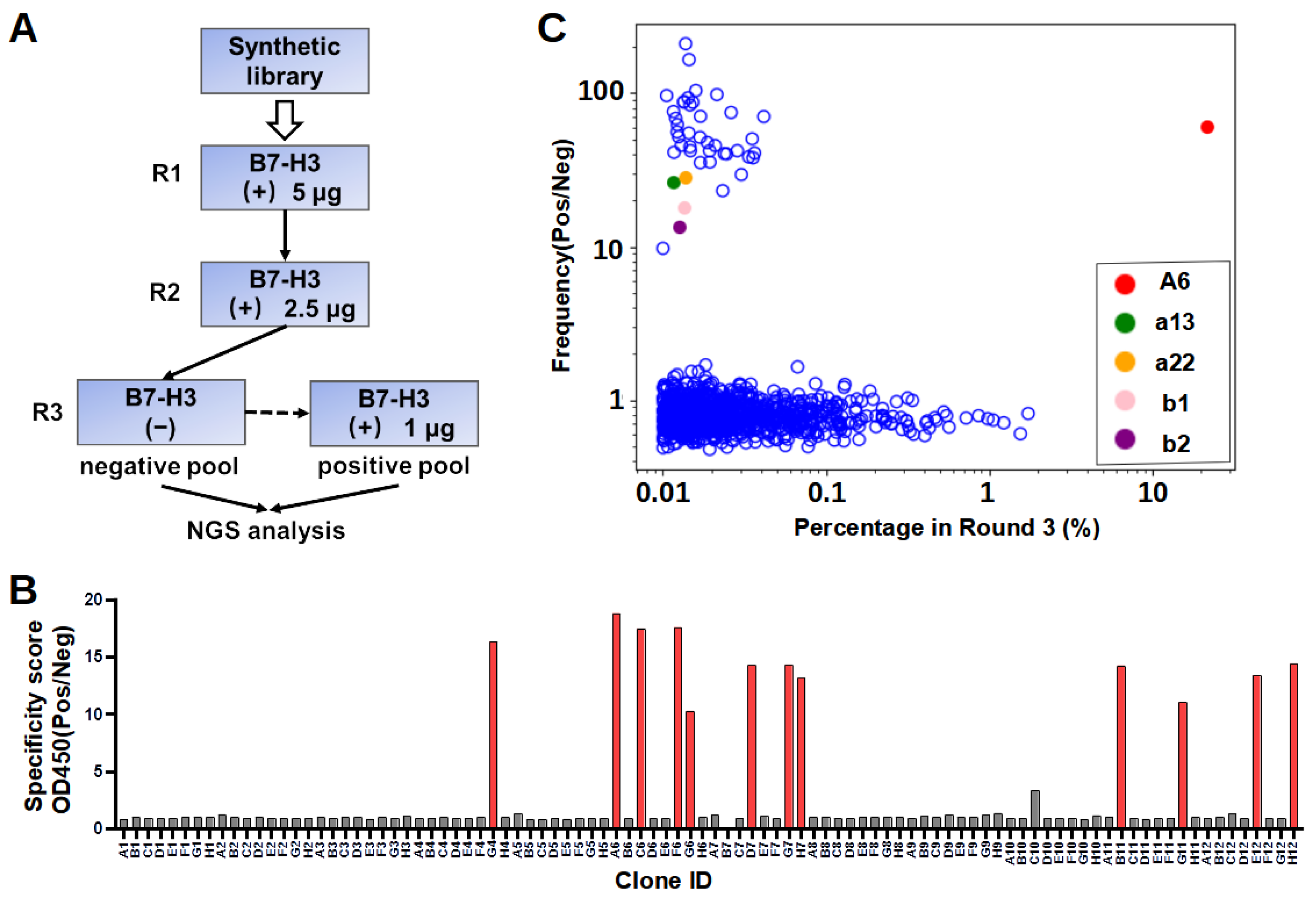
In our workflow of in vitro selection, the phage display library was counter-screened on blocked control wells to reduce the number of unspecific binders. The NGS of both the negative and positive pools enabled the calculation of the specificity scores to explore sequences with low abundance. It is noted that the lack of specificity for the majority of enriched sequences (
Figure 2C) is not uncommon, probably because the library was synthetic instead of immune, as observed in previous studies [
30,
31,
32]. In addition, the result showed two of the five selected sequences specifically bound to B7-H3. We also found that 36 clones identified by NGS demonstrated sequence similarities with A6, differing by only one to two amino acids. Similarly, a recent study reported that enriched clones with a high specificity were close homologs, sharing >80% identity in CDR L3 and H3 sequences [
30]. The success rate of binder identification based on the NGS specificity score was relatively low. This could be because the selected nanobody sequence adopted different conformations when recombinantly expressed compared to those in the pIII protein fusion form in phage surfaces. Sequencing errors cannot be excluded as one of the potential possibilities of mis-identification.
Recent studies have discovered that the location of epitopes have a significant impact on the mode of action and efficacy of monoclonal antibody [
25] and bispecific antibody [
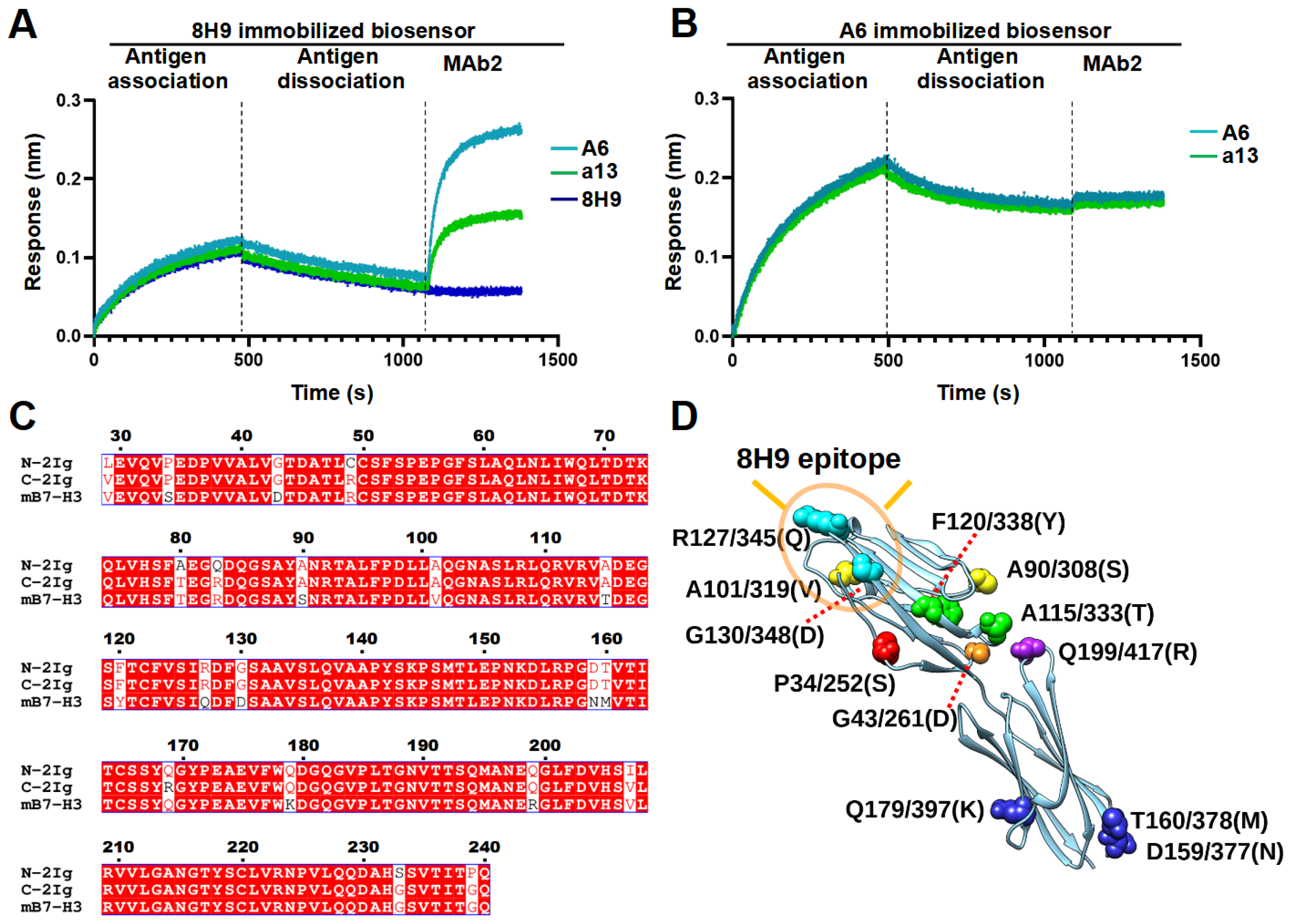
33]. Therefore, the development of various antibodies proves invaluable in pinpointing the potent epitope. B7-H3 possesses two distinct domains, namely, IgC and IgV. Although the epitope for most B7-H3 antibodies remains undescribed, two of the well-established clones, 8H9 and 376.96, were identified as binding to the FG loop in the IgV domain [
27,
28]. This loop has been previously associated with a possible role in inhibiting T cell proliferation [
34]. Li et al. (2023) recently generated dromedary camel nanobodies targeting the IgC domain of B7-H3 and demonstrated that CAR-T cells based on one of the nanobody sequence had potent anti-tumor activity against large tumors in a mouse model [
28]. Notably, a recent study also reported that murine-derived MAb 7C4 targeted an epitope in the IgC domain involving Q179, resulting in a human B7-H3-specific binding profile similar to A6 [
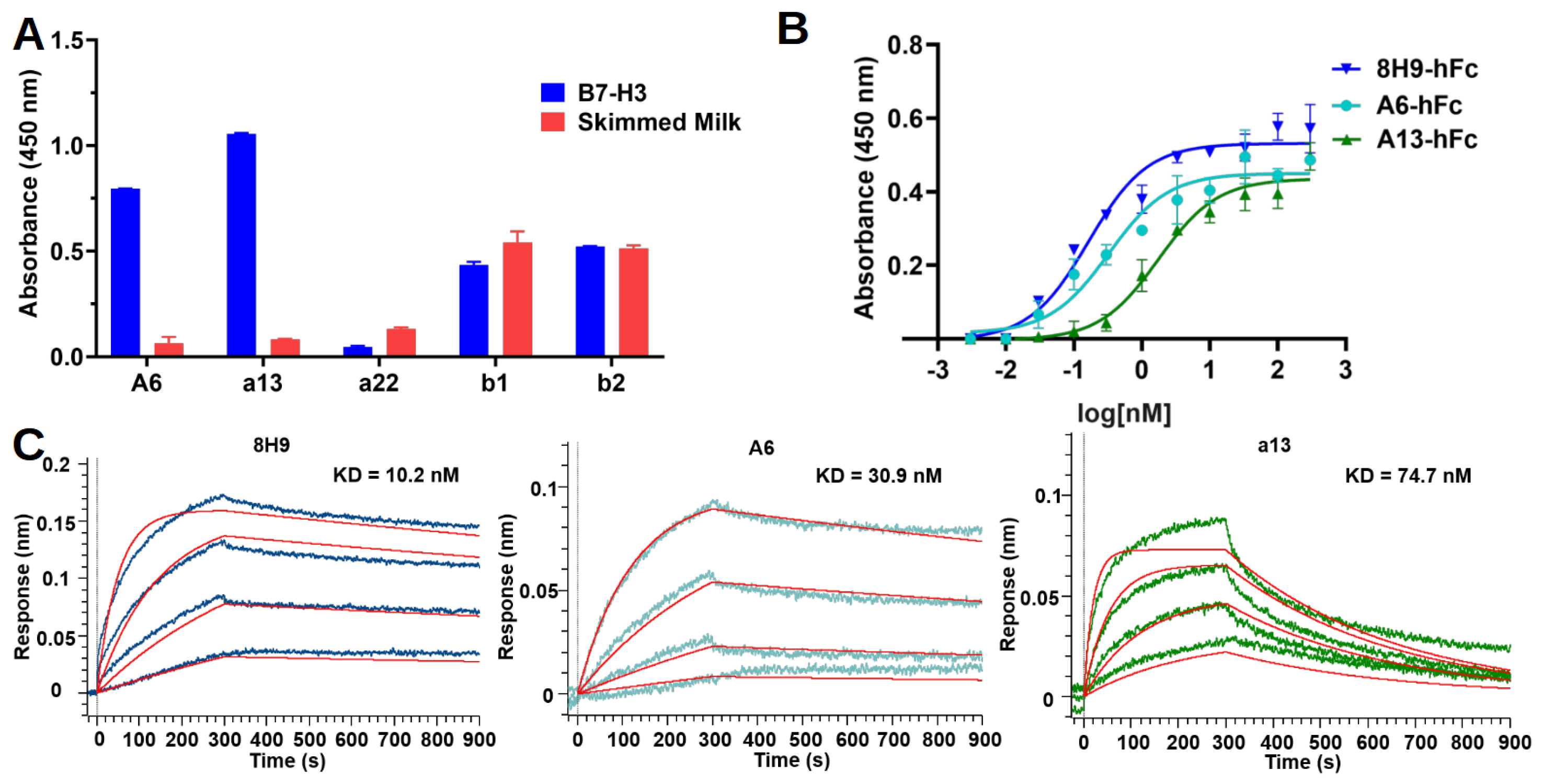
33]. In this study, we found that both A6 and a13 were bound to a distinct region compared to 8H9. However, the exact epitopes of these two nanobodies still await further study. Moreover, the potency of using these nanobodies in different formats of immune therapy, including MAbs, bispecific antibody, and CAR-T cell, will enable the understanding of the significance of targeting this epitope.
Anti-B7-H3 agents, including MAbs, bispecific antibodies, antibody-drug conjugates (ADCs), CAR-T cells, and radioimmunotherapy agents, have exhibited encouraging anti-tumor activity in preclinical models and have recently entered clinical trials for multiple cancer types [
11]. B7-H3-specific ADCs have been actively developed to treat solid tumor. For instance, pyrrolobenzodiazepine-conjugated B7-H3 ADCs killed both cancer cells and tumor vasculature, eradicating large established tumors and metastases in a preclinical study [
16]. DS-7300a (Daiichi Sankyo, Tokio, Japan), an ADC consisting of a B7-H3-specific MAb conjugated to four topoisomerase I inhibitor particles, is currently being tested in a Phase I/II trial [
35]. Additionally, MGC018 (humanized B7-H3 MAb with a cleavable linker-duocarmycin payload, MacroGenics, Rockville, MD, USA) entered a clinical trial based on encouraging data in a preclinical model [
36], although it has been terminated due to business reasons.
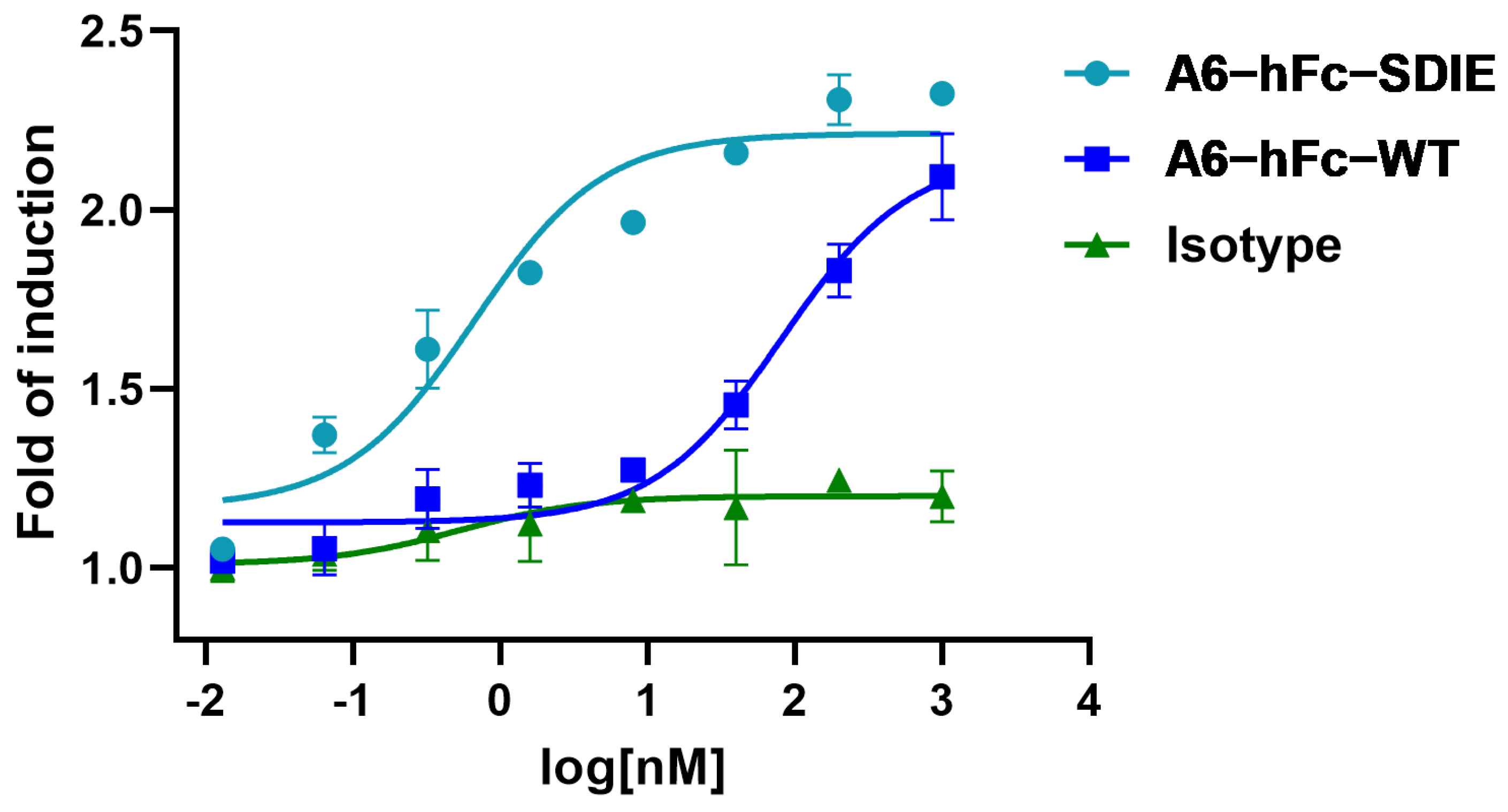
Another promising B7-H3-targeting immunotherapeutic strategy is represented by Fc-enhanced MAbs. Enoblituzumab (MGA271, MacroGenics, Rockville, MD, USA), a fully humanized MAb bearing an Fc domain engineered to enhance its anti-tumor function, was found in Phase I trials to be well-tolerated [
37]. Enoblituzumab is currently being evaluated in a Phase II trial against localized prostate cancer as neoadjuvant [
38]. The rationale for using an engineered Fc domain to improve the effector cell function has been supported by the correlation of clinical outcomes of MAb therapies with the natural polymorphisms of Fcγ receptors (FcγRs) in patients. In this study, A6-hFc showed a dose-dependent ADCC effect on B7-H3-expressing cells in the luciferase-based assay. It has been shown that the two point mutants (S239D/I332E) of the Fc domain used in this study enhanced effector-mediated anti-tumor functions via increased affinity for the activating receptor CD16A [
39]. Outstandingly, the A6 fused to this enhancing Fc variant further improved the ADCC activity by approximately 100 folds (
Figure 6). It is important to note that the current ADCC data were derived from in vitro reporter assays, and further investigation in in vivo models can validate the efficacy of A6 in these IgG formats.
Multispecific antibodies utilizing B7-H3-specific MAbs represent an additional B7-H3 targeting modality. For instance, anti-B7-H3 IgG has been fused to either an anti-CD3 or anti-4-1BB single-chain variable frament (scFv) to construct bispecific antibodies to recruit and activate T cells against cancer cells [
33,
40]. In another study, an anti-PD-L1 IgG fused to an anti-B7-H3 nanobody was shown to induce a synergetic effect [
41]. Anti-B7-H3 antibodies have also been linked to CD16-specific antibodies, which can lead to NK-cell-mediated cancer cell lysis [
42,
43]. Lastly, B7-H3-targeting CAR-T cells have been generated, and some of them are being evaluated in trials targeting different solid tumors [
28,
44]. The A6 clone is currently used as the B7-H3-specific arm to develop bispecific and trispecific antibodies in the lab. The synthetic library developed in this study can be further explored to discover nanobodies against different targets in order to ease the development of these multi-specific agents. Murine B7-H3-blocking MAbs were tested in mice, and the result showed that tumor growth reduced with CD8
+ T and NK-cells’ infiltration density increased [
45]. However, the limited information on the B7-H3 receptors hinders the translation of these findings to the clinical setting. Nanobodies discovered in this study can be tested for their ability to suppress human T-cell activation and validated as blocking antibodies to inhibit tumor growths in this context.
In summary, a large synthetic nanobody library was constructed in this study, with the desired CDR positions fully randomized using degenerate codons. This phage display library was successfully applied to isolate B7-H3-specific nanobodies, which were shown to target a distinct region compared to known MAbs. One of the clones, when fused to an enhancing Fc domain, showed a remarkable ADCC efficacy. This demonstrates that the synthetic libraries produced binders with functional activity. Future work will focus on the detailed epitope mapping of these newly isolated nanobodies and characterize their activity using both in vitro and in vivo models. The newly generated synthetic library is especially suited for nanobody discovery tasks where binder generation via immune libraries fails due to self-tolerance, toxic antigens, or insufficient stability of the antigen.
4. Materials and Methods
4.1. Construction of Synthetic Library
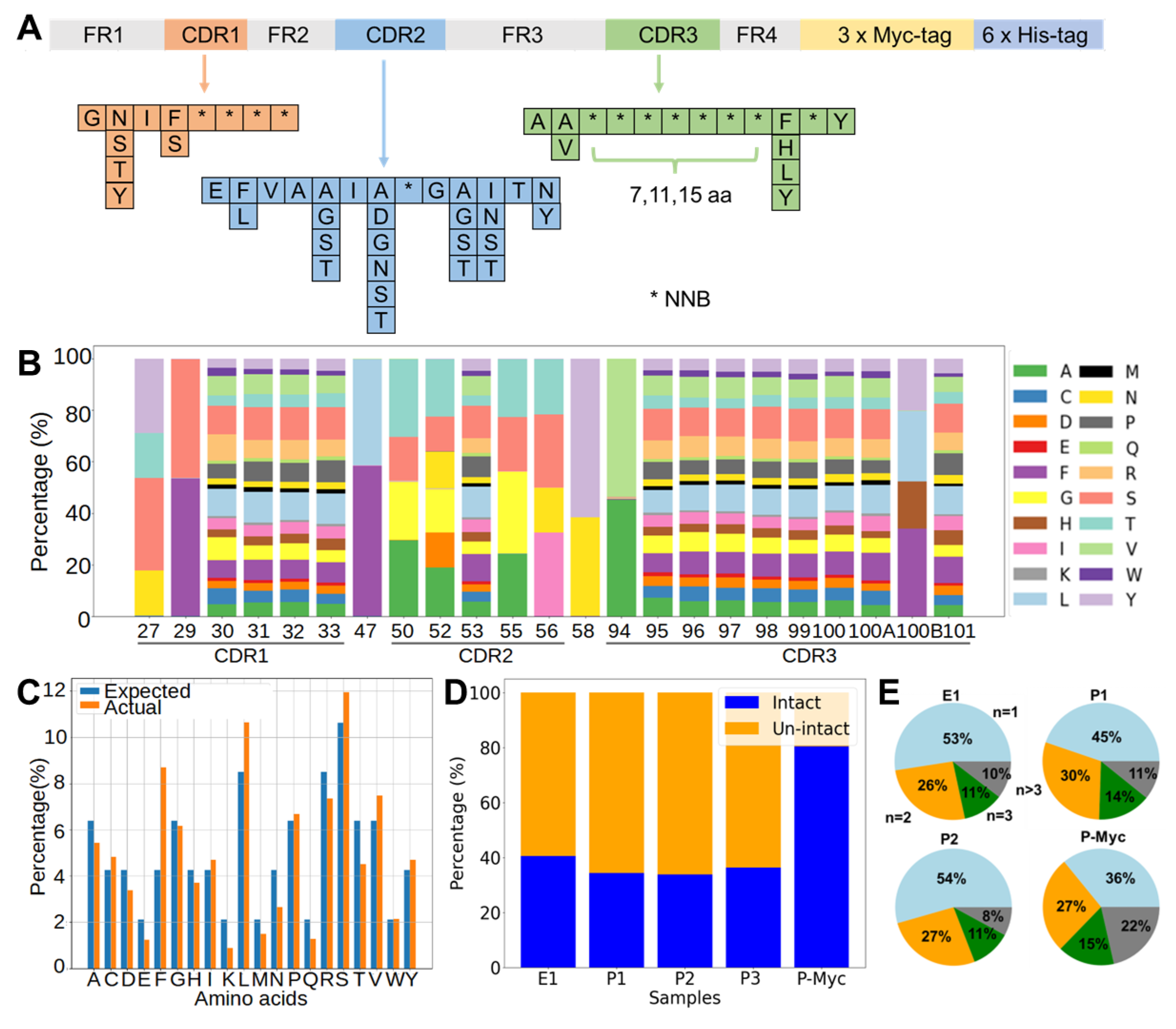
The DNA library of nanobodies was assembled by one-step assembly PCR, similar to a previous design [
4]. A set of ten primers, P1, P2, to P10 (
Table S1), were mixed to prepare “mix 9a”, “mix 9b”, and “mix 9c” containing each primer in an equimolar ratio, which only differed in the P9 primers (P9a, P9b, and P9c), to introduce CDR3 regions of variable lengths with 7, 11, or 15 continuous NNB codons. A concentration of each primer at 0.25 µM in the mixed pool produced optimal yields for the assembly PCR with a PrimeSTAR Master Mix (Takara Bio, Dalian, China). PCR products with the correct size were purified by DNA agarose gel extraction and used as templates for attaching a 3×Myc-tag by an overlapping PCR. The final products were purified and mixed in a 1:2:1 molar ratio of short/medium/long CDR3 loops. The mixture was digested with a PstI/BstEII restriction enzyme (NEB, Ipswich, MA, USA) and ligated with linearized pMES4 plasmids (GenBank GQ907248) using T4 DNA ligase (Takara Bio, Dalian, China). The ligation product was subsequently purified and transformed to an electro-competent
E. coli TG1 strain. Library diversity was estimated by plating serial dilutions of the transformed bacteria.
The transformed TG1 cells were plated on large agar plates. All colonies were scraped off the plates with a 5 mL 2 × Yeast Extract Tryptone (2×YT) medium. After complete resuspension, these TG1 cells were diluted in a 2×YT medium to a initial optical density at 600 nm (OD600) of 0.1 and grew until the OD600 reached 0.6–0.8. To amplify phages, the culture was mixed with helper phage M13KO7 (molar ratio of TG1:M13KO7 = 1:20) and incubated with 0.2 mM Isopropyl β-D-Thiogalactoside (IPTG) and 50 μg/mL Kanamycin with constant shaking overnight at 30 °C. The phage was harvested by precipitation with 20% polyethylene glycol (PEG) 8000/2.5 M NaCl. The precipitated phages were resuspended in a PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 1.8 mM KH2PO4, pH 7.4), aliquoted and stored at −80 °C for later use.
Unproductive sequences due to insertion, deletion, or frame shift can be reduced by one round of anti-Myc selection. Protein G magnetic beads (Invitrogen, Carlsbad, CA, USA) were coated with an anti-Myc antibody (clone 9E10) and washed three times with PBS containing PBST. The phage solution ( CFU) was pre-cleared with the Protein G magnetic beads. The supernatant was then transferred to 34 µL antibody-coated beads and incubated at room temperature for 30 min. The beads were washed three times with PBST and eluted with 50 µL 50 mM glycine (pH 2.5). Eluates were neutralized with 50 µL 1 M Tris-HCl (pH 7.5).
4.2. Next-Generation Sequencing for Nanobody Library
4.2.1. Amplicon Preparation for Sequencing
DNA from E. coli library was isolated using the plasmid extraction method. Then, CFU of E. coli was centrifuged at 10,000 rpm for 5 min and then extracted by a Plasmid Mini Kit (Omega Bio-Tek, Norcross, GA, USA). DNA from the phage library was isolated using the sodium iodide–ethanol precipitation method. The steps below were for 500 µL of solutions containing – CFU/mL of phages. Firstly, the phage solution was mixed with a 200 µL PEG/NaCl solution and incubated on ice for 2 h. The solution was then centrifuged at 12,000 rpm for 15 min at 4 °C, and the supernatant was discarded. The pellet was thoroughly dissolved in a 5 M sodium iodide solution (63 µL), 100% ethanol (156 µL) was subsequently added, and the mixture was further incubated on ice for 2 h to precipitate DNA. A second centrifugation (12,000 rpm, 4 °C, 15 min) was conducted to harvest DNA as a white or translucent pellet. Finally, the pellet was resuspended in 70% ethanol (200 µL) to remove residual salts. The solution was centrifuged at 12,000 rpm at 4 °C for 15 min, and the ethanol supernatant was discarded. The pellet was dried for 20 min at room temperature.
The extracted DNA was used for PCR amplification with primers flanking the variable region (10 µL PCR reaction containing 40 ng DNA template, 15 cycles) (
Table S1). The PCR product was examined in a 1.5% (
w/
v) agarose gel with Tris-acetate-EDTA buffer. The band corresponding to the expected product was extracted from the gel and purified using an E.Z.N.A.
®Gel Extraction Kit (Omega Bio-Tek, Norcross, GA, USA). Purified amplicons were sent for sequencing library preparation and NGS in GENEWIZ company (Suzhou, China). NGS was performed with Novaseq paired-end 2 × 250 bp (Genewiz, Suzhou, China). At least one million reads were collected for each sample.
4.2.2. NGS Data Analysis
Paired-end reads were merged to generate complete sequences according to the overlap regions between Read1 and Read2 using FLASH [
46]. The adaptor and primer sequences were trimmed using Cutadapt [
47]. The Seqkit tool set was used to translate and analyze the sequences [
48]. Sequences were filtered to generate intact sequences by estimated amplicon lengths, correct amino acids at the end of the sequencing region, and absence of early stop codon. The percentage of intact sequences was calculated as the number of intact sequences divided by the total number of successfully merged and trimmed sequences. Python scripts, which were in-house written and will be made available upon request, were used to calculate (1) the percentage of each amino acid type in a specific position, (2) the percentage of each amino acid type averaged over all fully randomized sites in the nanobody library, and (3) numbers of each unique sequence in the nanobody library. The figures were created using python’s matplotlib [
49].
4.3. Expression and Purification of B7-H3
Human 4IgB7-H3 and murine 2IgB7-H3 ectodomain were subcloned from a gene vector (Sino Biological, Beijing, China) into the pcDNA3.1(+) expression vector. The expression vectors were chemically transformed into DH5a E. coli. For expressing human 4IgB7-H3 ectodomain, 200 μg endotoxin-free expression plasmids were transfected to 200 mL Expi293F cells (Thermo Fisher, Walthem, MA, USA) with a Polyethylenimine MAX (PEI MAX) transfection reagent (Polysciences, Warrington, PA, USA). Supernatants from transfected cells were collected after 96 h and then filtered through a 0.22 µm Polyethersulfone (PES) filter (NEST, Wuxi, China). The filtered supernatant was mixed with equal volumes of a binding buffer (20 mM NaH2PO4, 20 mM Na2HPO4, 500 mM NaCl, 10 mM imidazole, pH 7.4) and purified using HisTrap HP column (Cytiva, MA, USA) in an AKTA Pure protein purification system. Subsequently, the column was washed with the binding buffer until a stable baseline was attained and eluted with an elution buffer (20 mM NaH2PO4, 20 mM Na2HPO4), 500 mM NaCl, 500 mM imidazole, pH 7.4). The eluate was concentrated using a Vivaspin centrifugal concentrator (Sartorius, Göttingen, Germany) with a molecule weight cut-off (MWCO) of 30 killo-Dalton (kDa) for human B7-H3 and 10-kDa for murine B7-H3. After the proteins were dialyzed to a PBS buffer, a final polishing of the proteins was conducted using size exclusion chromatography (Superdex 200 10/300 GL). The soluble B7-H3 protein was mixed with a final concentration of 50% glycerol and stored at −80 °C. To analyze the binding epitope, N-terminal 2Ig (residue 29–246) and C-terminal 2Ig (residue 247–458) of human B7-H3 ectodomain were subcloned from a gene vector (Sino Biological, Beijing, China) into the pcDNA3.1(+) expression vector. The expression and purification process was similar to the process described above.
4.4. Phage Display Panning
Recombinant 4IgB7-H3 proteins were coated onto a 96-well plate (Thermo Fisher, Walthem, MA, USA, 439454) for phage display panning. Briefly, five wells were each coated with 250 ng of recombinant proteins in PBS overnight at 4 °C. All wells were then washed with PBST three times and blocked with a blocking buffer (PBS with 5% skimmed milk) at room temperature for 1 h. Subsequently, – CFU of blocked phages were added to each well and allowed to bind for 1 h at room temperature. After incubation, the wells were washed 15 times with PBST and 5 times with PBS to clear unbound phages. Finally, 100 µL of 100 mM glycine (pH 2.5) was added to each well and incubated at room temperature for 15 min to elute bound phages. The eluate was immediately neutralized by a 50 µL 1 M Tris-HCl buffer (pH 7.5). A small fraction of the neutralized eluate was serially diluted with a 2×YT medium and used for titer measurements. To produce phages for further panning, the TG1 cells (OD = 0.6–0.8) were infected by the eluate with constant shaking for 60 min and were spread in large agar plates with 100 μg/mL ampicillin for overnight incubation at 30 °C. Phages were packaged from TG1 cells and purified as described above. In the third round, the amplified phages from the second round were first incubated with skimmed-milk-blocked control wells. Unbound phages were then transferred to the B7-H3 protein-coated wells for panning. The washing, elution, and amplification steps were similar to the previous panning steps.
4.5. Phage ELISA
After three rounds of selection, recovered TG1 cells were plated, and colonies were randomly picked to prepare single-clone phages for ELISA. In brief, individual colonies were picked and diluted to a 200 µL 2×YT medium with ampicillin in a 96-cell culture plate for overnight growth at 37 °C. The overnight culture in each well was diluted 100 times to a fresh medium and continued incubation at 37 °C. M13KO7 was added when OD600 reached 0.6–0.8. After incubating at 37 °C for 30 min, phage production was induced by the addition of IPTG (0.2 mM) and further incubation at 30 °C for 16 h. In ELISA, each well in the Maxisorp plate (Thermo Fisher, Walthem, MA, USA) was coated with recombinant proteins or a skimmed milk control (250 ng/well) in a PBS buffer. After being blocked with 3% bovine serum albumin (BSA) (Sigma, St. Louis, MO, USA) in PBS, 80 µL of phages were transferred to each well and incubated at room temperature for 2 h. The plates were washed with PBST five times and then incubated with Horseradish peroxidase (HRP) conjugated Anti-M13 antibody (Sino Biological, Beijing, China, 11973) at room temperature for 1 h. Subsequently, the plates were washed with PBST five times and developed by incubating with a tetramethylbenzidine (TMB) substrate (GBCBIO, Guangzhou, China) (50 µL/well) for 3 min. The reaction was quenched by adding 50 µL of 1 M HCl. Absorbance at 450 nm was measured with Multiskan FC microcoder (Thermo Fisher, Walthem, MA, USA).
4.6. Expression and Purification of Nanobody-hFc
The nanobody A6 and a13 sequences were subcloned to pFUSE-hIgG1-Fc2 (InvivoGen, San Diego, CA, USA), which was modified to insert a 6xHis-tag at the C-terminal of the Fc domain, to produce a nanobody–human IgG1 Fc fusion antibody (Nanobody-hFc). An Fc-enhanced A6 antibody was constructed to include two mutations in the Fc domain, specifically serine (S) at residue 239 to aspartic acid (D), and isoleucine (I) at position 332 to glutamic acid (E). This mutant was named A6-hFc-SDIE. The nanobody-hFc was produced by an Expi293F transient expression system (Thermo Fisher, Walthem, MA, USA) as described above for recombinant B7-H3. Samples were purified using TALON resin (Takara Bio, Dalian, China) and polished by size exclusion chromatography (Superdex 200 10/300 GL). Purified antibodies were concentrated and buffer exchanged to PBS.
4.7. Nanobody-hFc ELISA
Nanobody-hFcs were tested for binding in ELISA against recombinant human 4IgB7-H3. Briefly, protein and skimmed milk control were coated on the Maxisorp plate as described above. On the next day, the plate was blocked with 3% BSA in PBS. The antibodies were diluted with a blocking buffer to 50 nM and added to wells to incubate for 1 h at room temperature. The plate was washed five times with PBST and then incubated for 1 h with an HRP-conjugated goat anti-human IgG-Fc secondary antibody (Proteintech, Chicago, IL, USA, SA00001) diluted 1:5000 in the blocking buffer. After five washes with PBST, the plate was developed as described above for the phage ELISA. In the dose-dependent ELISA experiment, three-fold serial dilutions of antibodies (starting from 300 nM) were added to the plate in duplicates for each concentration of one antibody.
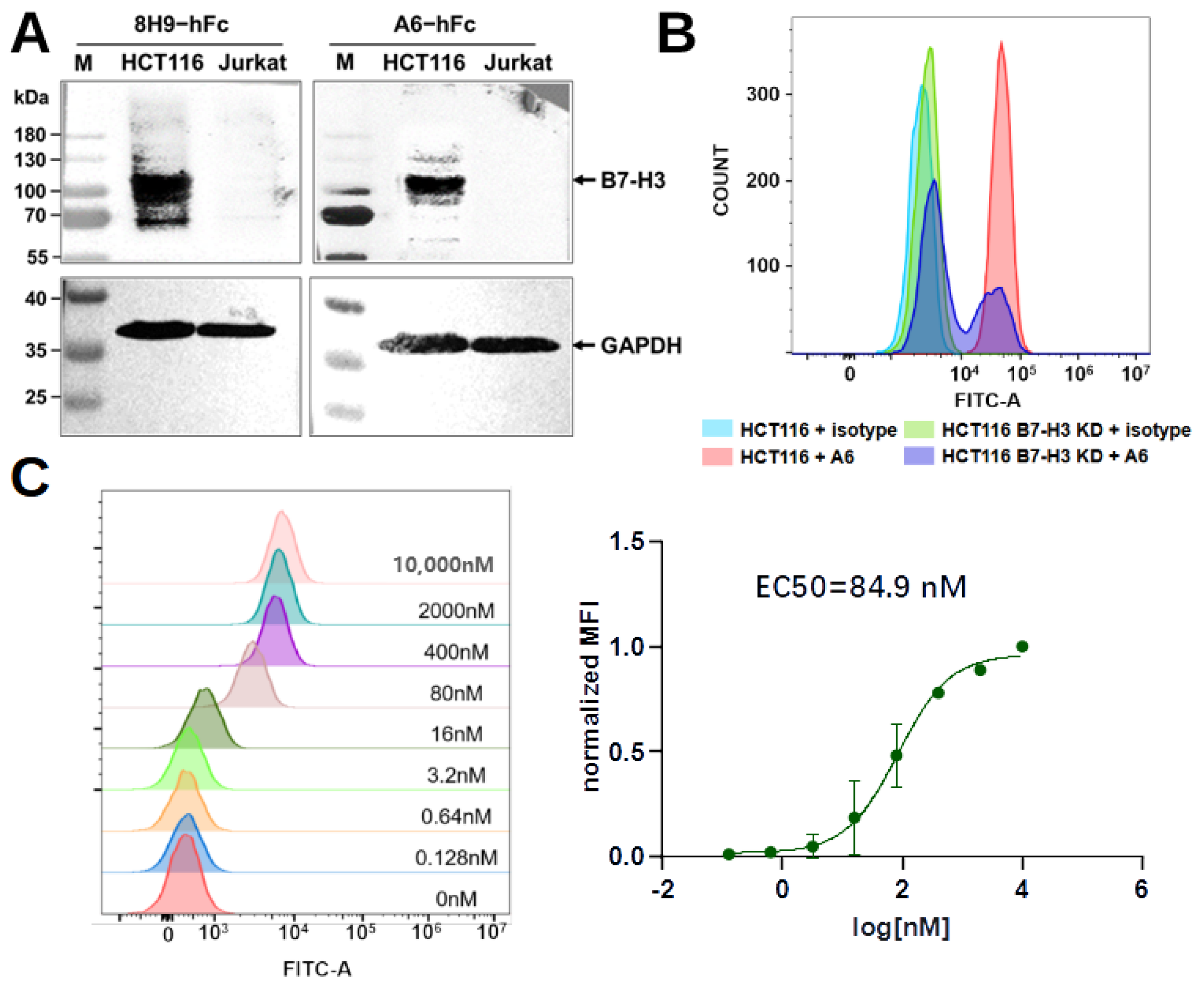
4.8. Western Blot
The preparation of HCT116 or Jurkat cell (ATCC) lysate for sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE): cells were collected with the medium removed and diluted in a 200 µL lysis buffer (150 mM NaCl, 50 mM Tris, 1% TritonX-100, 1% phenylmethylsulfonyl fluorid, pH 8.0). After incubation for 30 min, the sample was centrifuged at 12,000 rpm for 10 min to collect the supernatant. The samples were separated on a 10% SDS-PAGE and transferred to Polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5% skimmed milk diluted in PBST overnight at 4 °C and then incubated with A6-hFc (200 nM) for 1 h at room temperature. The membranes were then washed five times with PBST and incubated with an HRP-conjugated goat anti-human IgG-Fc secondary antibody (Proteintech, Chicago, IL, USA, SA00001) diluted 1:5000 in the blocking buffer. For loading control, we used a rabbit monoclonal anti-GAPDH antibody (HuaBio, Hangzhou, China, HA721136) at 1:10,000 dilution as the primary antibody and an HRP-conjugated mouse anti-rabbit IgG (Santa Cruz, Dallas, TX, USA, 1:5000) as the secondary antibody. Detection was performed using Omni-ECL femtolight Substrate (EpiZyme, Shanghai, China, SQ202).
4.9. Generation of B7-H3 Knockdown HCT116 Cells
A Cas9 sgRNA plasmid system was used to generate B7-H3 knockdown HCT116 cell pools. A B7-H3 sgRNA target sequence was designed by the gRNA design tool (
genscript.com, accessed on 1 June 2023). An sgRNA sequence 5’-TTGATGTGCACAGCGTCCTG-3’ was chosen and cloned into pSpCas9(BB)-2A-Puro (PX459) V2.0 (Tsingke Bio, Beijing, China). Endotoxin-free plasmid (2 μg) was used to transfect 2 mL of HCT116 cells in a six-well cell culture plate with a 6 μg PEI MAX transfection reagent (Polysciences, Warrington, PA, USA). After 48 h, the supernatant was aspirated out, and a fresh culture medium with 2 μg/mL puromycin (BioFroxx, Einhausen, Germany) was added. The cells were incubated for three days under antibiotic pressure. Cells after this antibiotic-resistance screening were transferred to a new plate for expansion and protein expression detection. To detect protein expression,
cells were collected to prepare whole cell lysates as described above and quantified by a BCA Protein Assay Kit (CWBio, Beijing, China). Lysates containing 30 μg of proteins were run on 10% SDS-PAGE and transferred to a PVDF membrane. The membrane was blocked with 5% skimmed milk in a TBST buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 0.1% Tween-20) overnight at 4 °C and then incubated with a rabbit polyclonal anti-B7-H3 antibody (Sino Biological, 201526) at 1:1000 dilution, or rabbit monoclonal anti-GAPDH antibody (HuaBio, Hangzhou, China, HA721136) at a 1:10,000 dilution for 1 h at room temperature. An HRP-conjugated mouse anti-rabbit IgG (Santa Cruz, 1:5000) was used as a secondary antibody. The membranes were washed five times with a TBST buffer for 5 min at room temperature. Lastly, the blot was developed by incubating it with an Omni-ECL femtolight Substrate (Epizyme, Shanghai, China).
4.10. Flow Cytometry
Flow cytometry was used to assess the binding of A6-hFc to HCT116 or B7-H3 knockdown HCT116 cell pools. A human IgG1, kappa isotype (HG1K-500, Sino Biological), served as the negative control. About cells were incubated with 100 nM A6-hFc or isotype control in the dark at 4 °C for 30 min. Cells were washed three times with ice-cold PBS buffer (pH 7.4) and then incubated with an FITC F(ab’)2 goat anti-human IgG Fcγ antibody (Biolegend, San Diego, CA, USA, 398006) in the dark at 4 °C for 30 min. Finally, the sample was analyzed with CytoFLEX (Beckman Coulter, Indianapolis, IN, USA).
For dose-dependent analysis, HCT116 cells () were incubated with a 5-fold serial dilution (from 10 μM) of A6-hFc in the dark at 4 °C for 30 min. The cells were washed, incubated with the secondary antibody, and analyzed as described above. The binding activity was represented using normalized mean fluorescence intensity (MFI). The MFIs of each sample were normalized by subtracting the MFI of the negative control and dividing by the difference between the MFI of the antibody at 10 µM and the MFI of the negative control.
4.11. Binding Kinetics with Biolayer Interferometer
Antibodies were diluted in a PBS buffer to a final concentration of 5 μg/μL. The antibodies were then incubated with EZ-Link Sulfo-NHS Biotin (Thermo Fisher, Walthem, MA, USA) in a molar ratio of 20:1 (biotin:antibody) on ice for 2 h. Free biotins were removed by centrifugation using Vivaspin centrifugal concentrator (Sartorius, MWCO 5000). The biotinylated antibodies were buffer-exchanged into PBS at a pH of 7.4.
Binding kinetics were studied using the Octet R8 system (Sartorius, Göttingen, Germany). Bindings were performed at 16 °C with shaking at 1000 rpm in a black 96-well plate (Greiner Bio-One, Kremsmünster, Austria) containing 200 μL of solution in each well. A PBST buffer (pH 7.4) containing 10 mg/mL BSA was used for the analyte dilution and washing. Streptavidin (SA) biosensors were rehydrated, equilibrated, and then loaded with biotinylated antibodies (50 nM) for 300 s to reach a shift at around 1.5 nm. The sensors were washed, associated with a three-fold serial dilution of antigens for 300 s, and allowed for antigen dissociation in running buffer for 600 s. Regeneration was repeated three times, with a short incubation (5 s) in glycine (pH 2.5) and neutralization in running buffer (10 s). An empty biosensor was used as a reference. The data were analyzed using Octet BLI Analysis 12.2 software. The sensorgrams were subtracted from the reference well, and the kinetics were analyzed using association and dissociation steps and fitted into a standard 1:1 binding model.
4.12. Epitope Binning
The SA sensors were equilibrated to reach baseline and then loaded with biotinylated antibodies (MAb1) at 50 nM, followed by a 180 s wash. Subsequently, the sensors were bound by antigens at 600 nM for 300 s and allowed for dissociation for 600 s. Finally, the sensors were allowed for the binding of the unbiotinylated second antibody (MAb2, 50 nM) for 300 s. The unbiotinylated form of MAb1 was used as a blocking control at the MAb2 stage to demonstrate complete blocking with a fully overlapping epitope.
4.13. ADCC Luciferase Reporter Assay
ADCC luciferase reporter assays were performed using Jurkat-Lucia NFAT-CD16 reporter cells (InvivoGen, San Diego, CA, USA), which are effector cells that stably express the FcγRIII receptor and an NFAT-response element-driving expression of luciferase. The ADCC assay buffer contained an RPMI 1640 medium (Thermo Fisher, Walthem, MA, USA) with a 10% fetal calf serum (Nobimpex, Herbolzheim, Germany), supplemented with penicillin and streptomycin (Biological Industries, Göttingen, Germany). HCT116 cells were diluted in the ADCC assay buffer and added to the cell culture plate for 20,000 cells per well, incubated for 24 h at 37 °C with 5% CO2. On the next day, supernatants in the well were removed and replaced with 90 µL of the fresh assay buffer. Five-fold serial dilutions (starting from 10 μM) of antibodies were added to each well (20 µL/well) and incubated for 1 h at 37 °C with 5% CO2. A human IgG1 kappa isotype (HG1K-500, Sino Biological) served as the negative control. After that, Jurkat-Lucia NFAT-CD16 reporter cells were added (90 µL/well) to a 20:1 effector/target cell ratio. The plate was incubated for 24 h and then centrifuged at 4500 rpm for 10 min. Supernatants (20 μL/well) were transferred to a 96-well white plate and then mixed with 50 μL of QUANTI-Luc™ 4 Reagent (InvivoGen, San Diego, CA, USA). The signal was measured immediately with the LUMINESCENCE program of the GloMax DISCOVER instrument (Promega, WI, USA). The data were fitted to a nonlinear model (three parameters) using GraphPad Prism 8 software.
4.14. Sequence and Structural Analysis
Sequence alignment was conducted using ESPript 3.0 [
50]. The predicted structure of the N-terminal 2Ig of human B7-H3 was extracted from an AlphaFold protein structure database [
51]. The B7-H3 protein expression patterns in different cell lines were extracted from the Human Protein Atlas [
52].



 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}