


Meta-Analytic Gene-Clustering Algorithm for Integrating Multi-Omics and Multi-Study Data





Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
- -
- Early integration which consists of concatenating all the omics data to one dataset then applying a single-omics clustering;
- -
- Late integration which consists of integrating the clusterings obtained after applying single-omics clustering to each omics;
- -
- The similarity-based methods which consist of computing similarity among sample for each omics, and then integrate them;
- -
- The dimension reduction-based methods;
- -
- The correlation and covariance-based methods which include the canonical correlation analysis (CCA) and the partial least square (PLS).
2. Methods
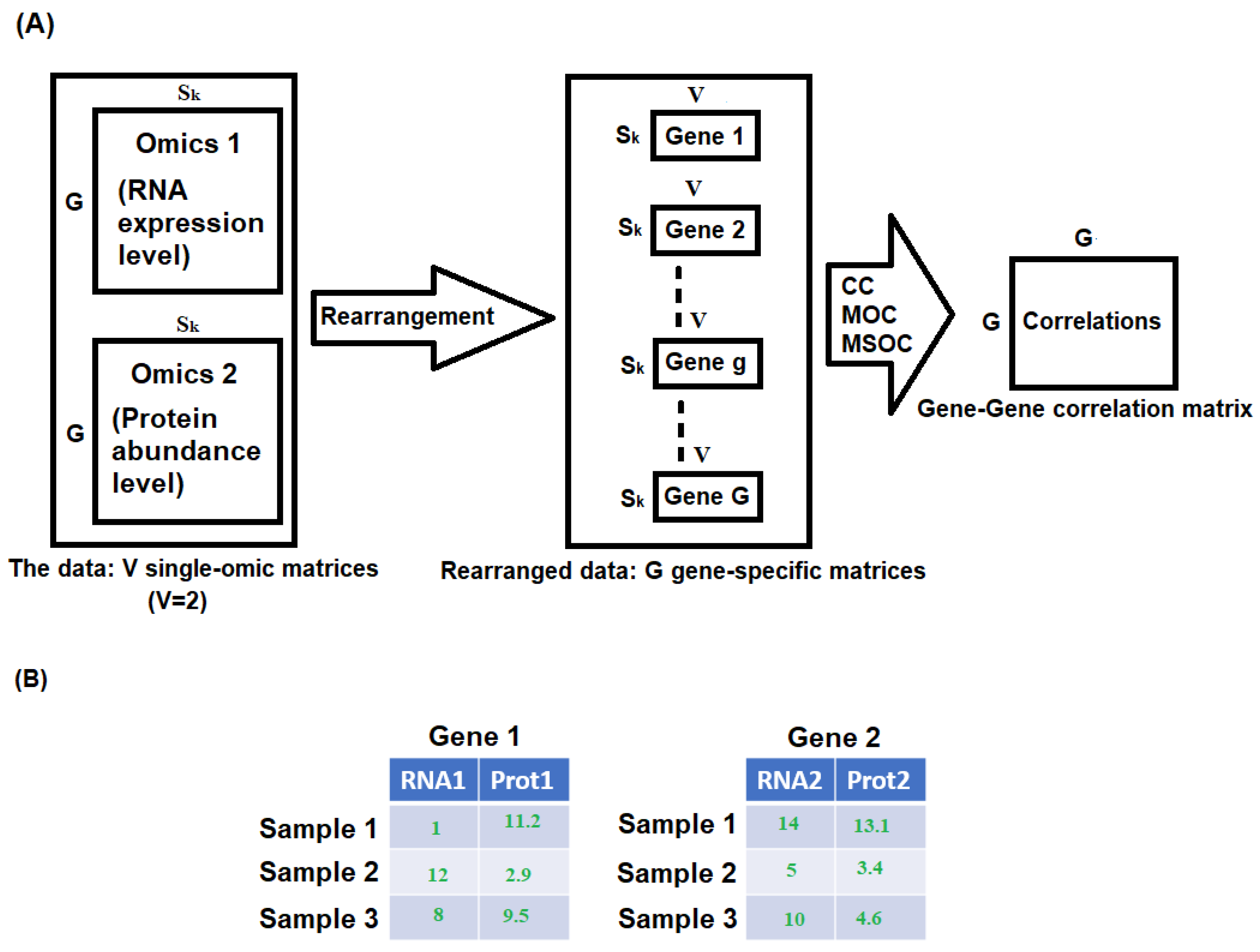
2.1. Step I. Computing the Gene-To-Gene Correlation for Each Study Using Multi-Omics Data
2.2. Step II. Combining the Within-Study Multi-Omics Gene-To-Gene Correlations from Multiple Studies
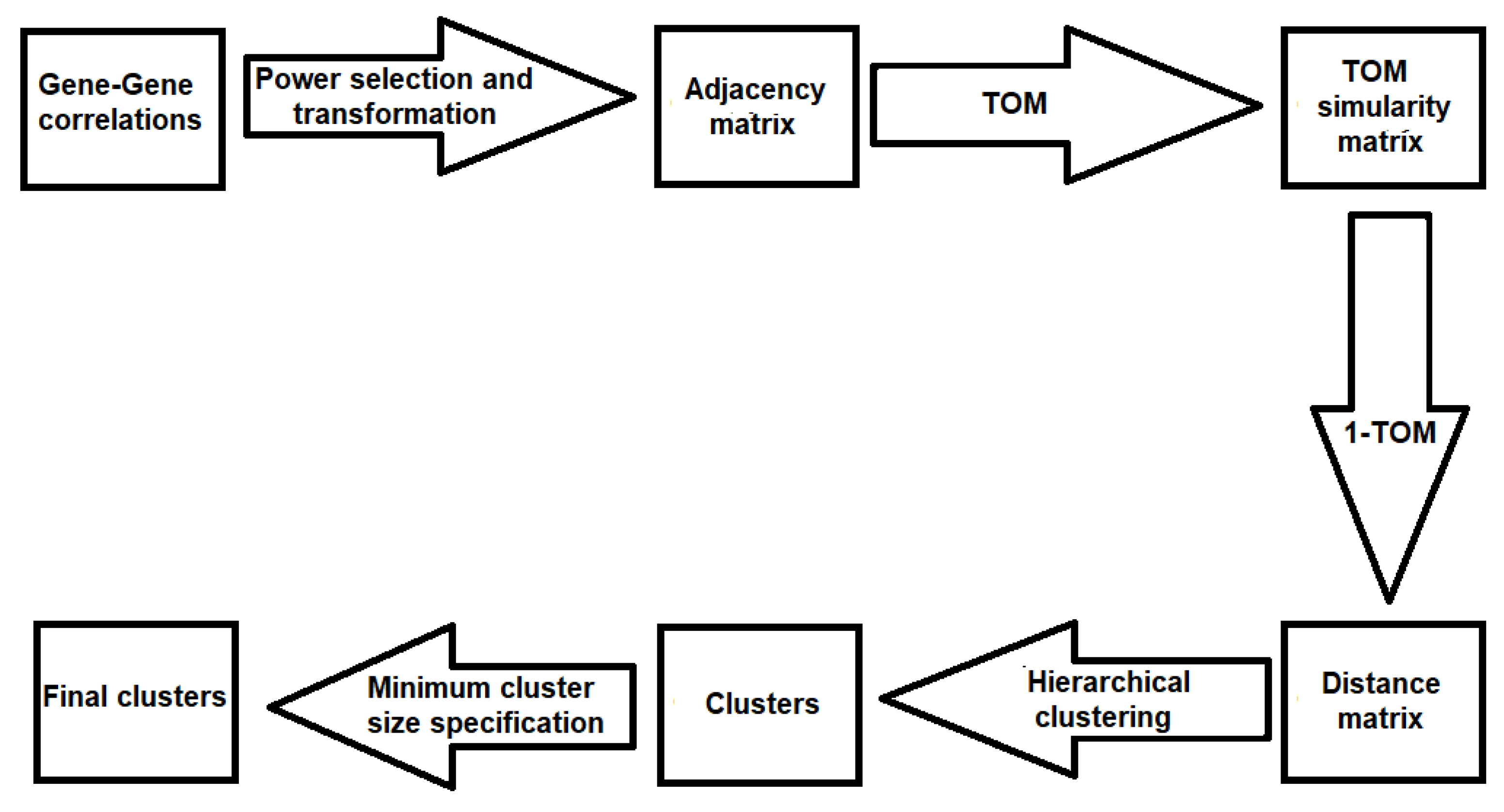
2.3. Step III. Gene Clustering Using the Modified WGCNA
2.3.1. Power Transformation
2.3.2. Computation of the TOM Similarity Matrix
2.3.3. Hierarchical Clustering
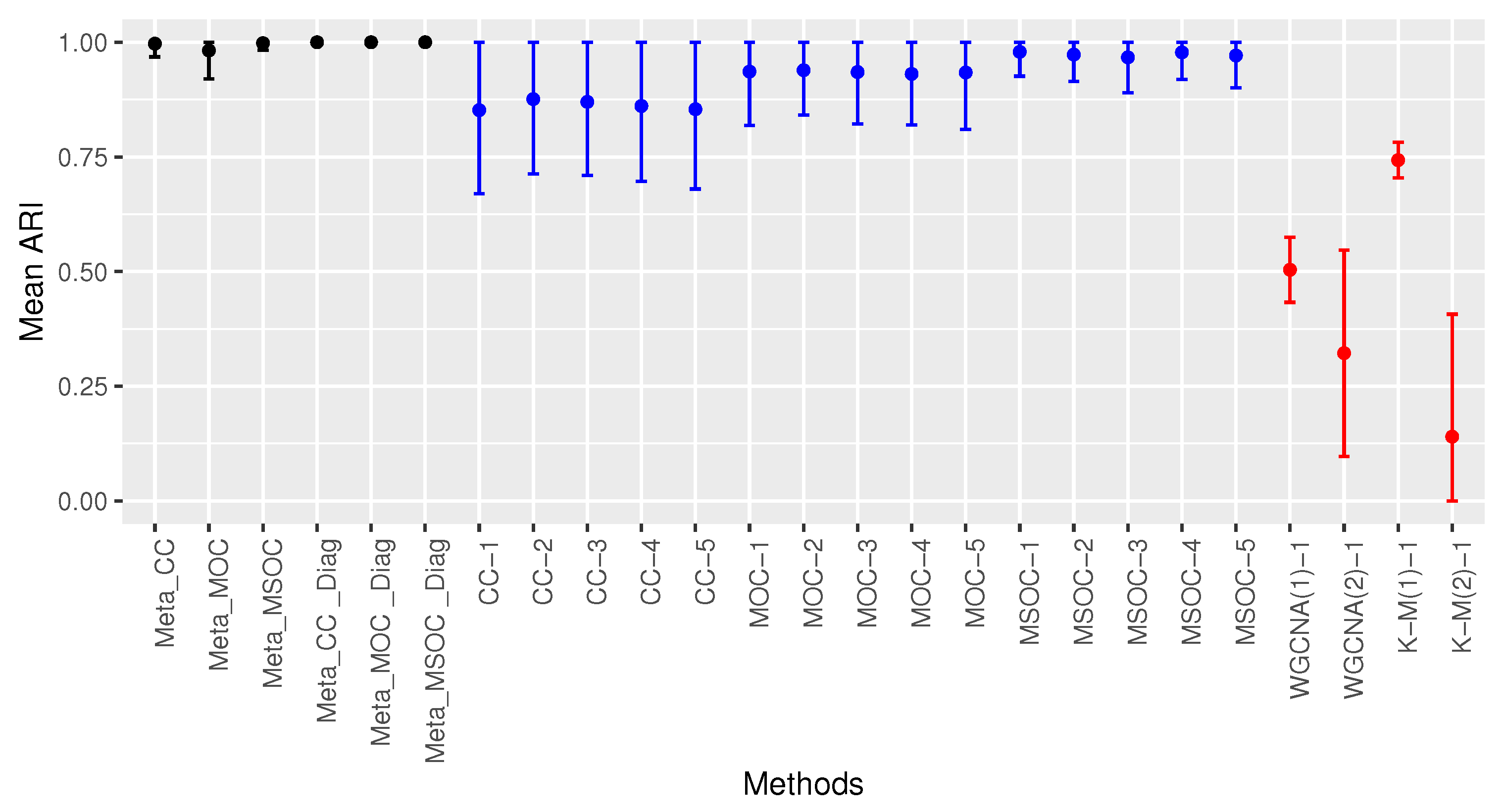
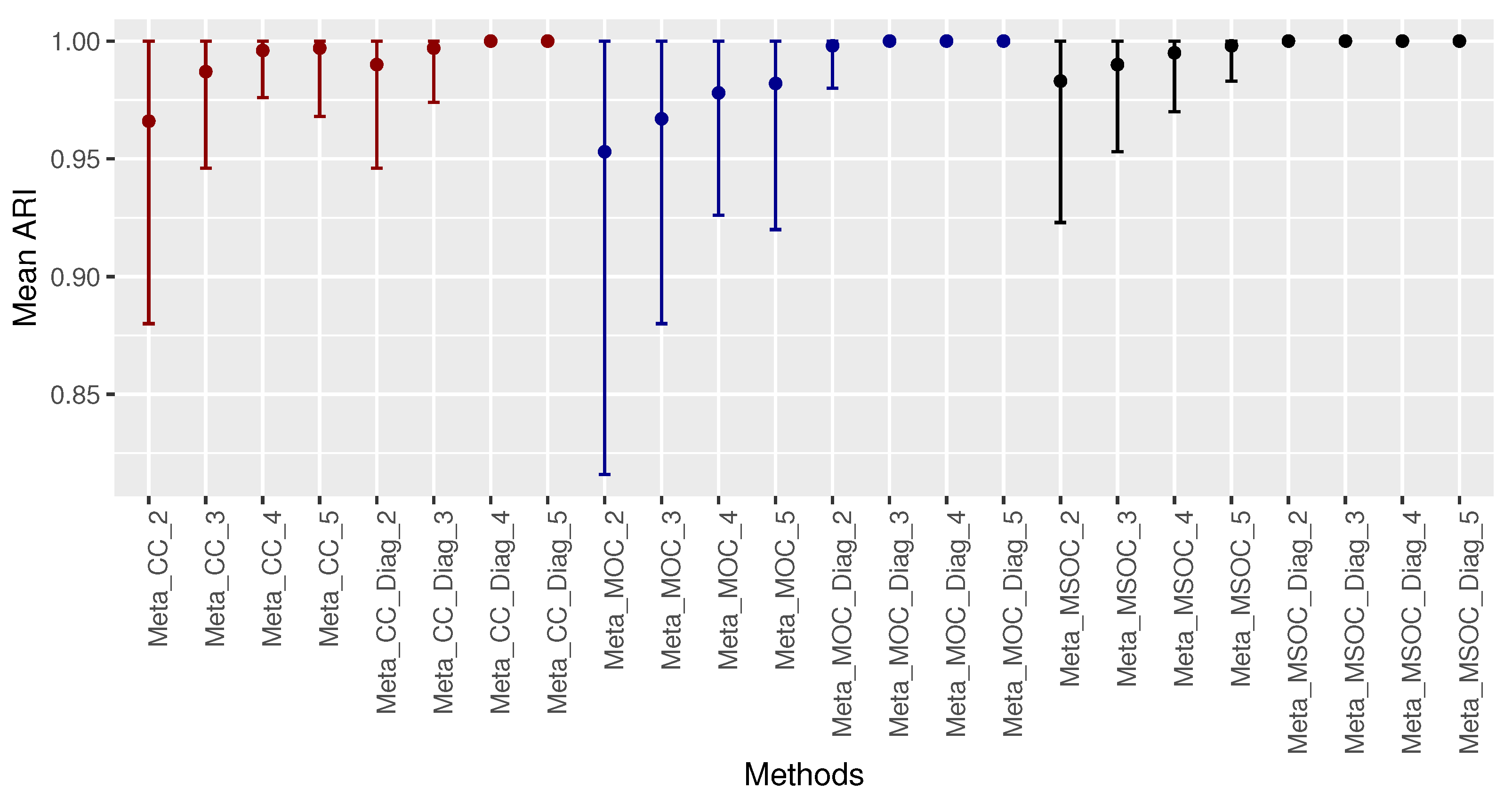
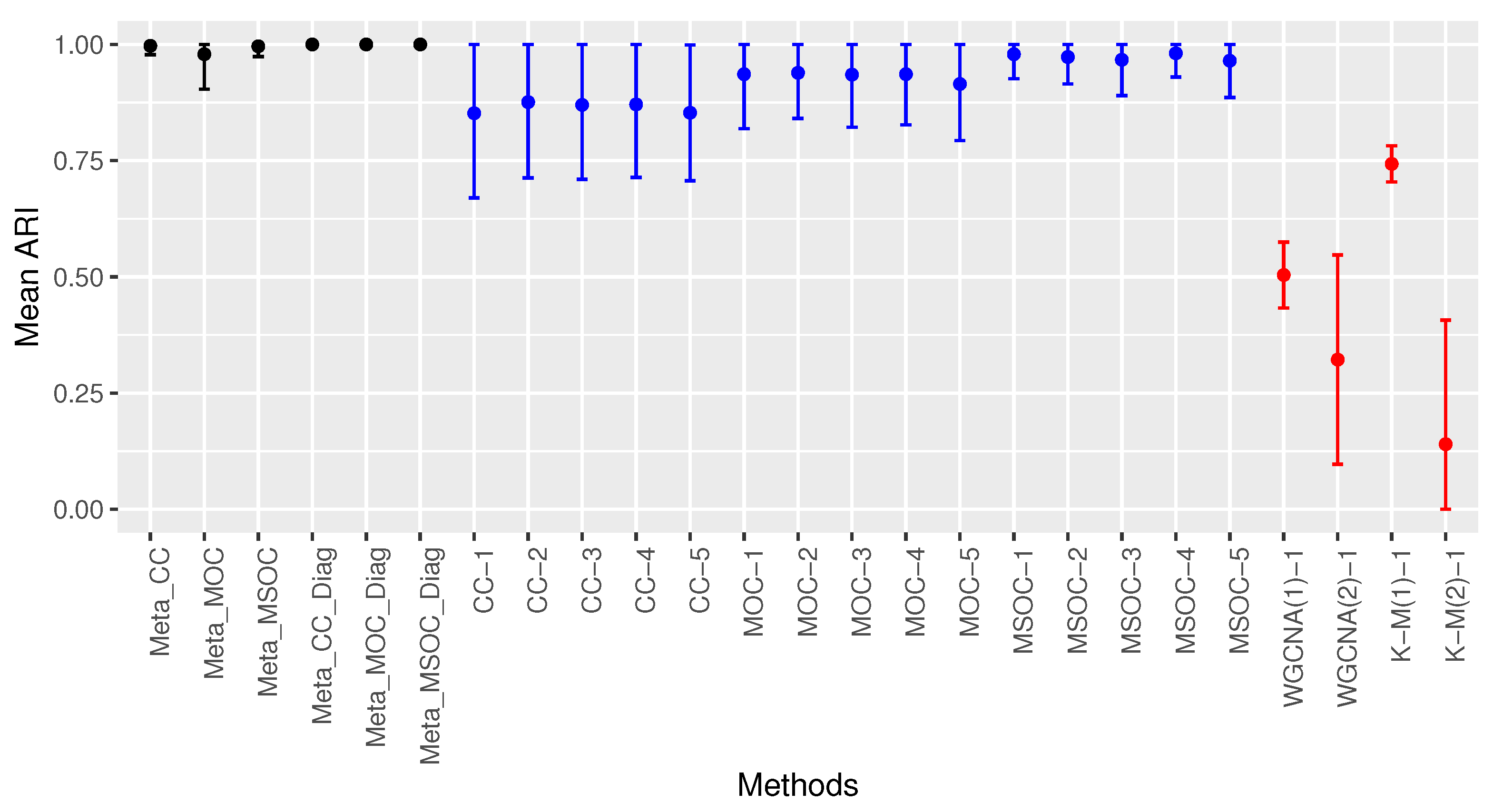
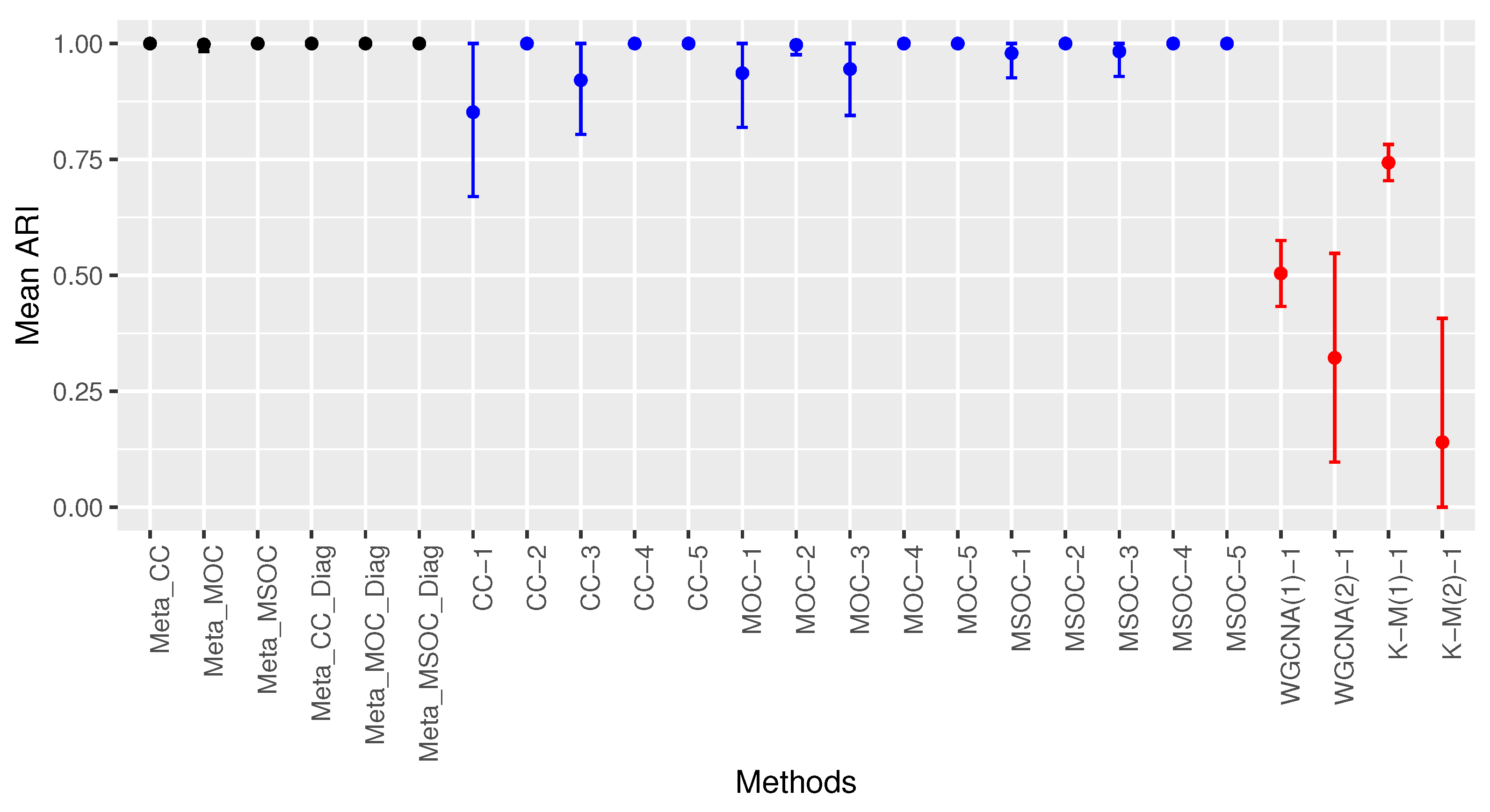
3. Simulation and Results
- Correlation between the omics within the same gene: ;
- Correlation between the same type of omics of different genes in the same cluster: ;
- Correlation between different types of omics of different genes in the same cluster: ;
- Correlation between omics from different clusters: .
- The modified WGCNA where the first power, which maximizes or minimizes the rate of change of the mean connectivity change rate, was selected: WGCNA( 1);
- The commonly used WGCNA where the power selected is the one that guarantees at least 60% adjusted goodness of fit while having a mean connectivity change rate of at most 1/3: WGCNA(2);
- The K-means method [17] with the true number of clusters c: K-M(1). It was run 25 times where the c starting points were randomly chosen and the best result was picked, based on minimizing the within-cluster variance. In the real world, the true value of c is unknown, so this method is favored compared to the other single-omics methods we examined;
- The K-means method where the gap statistic [18] was used to choose the value of c: K-M(2). The value of c was chosen from 1 to the true value of c plus 10. This was also run 25 times and the best result was picked.
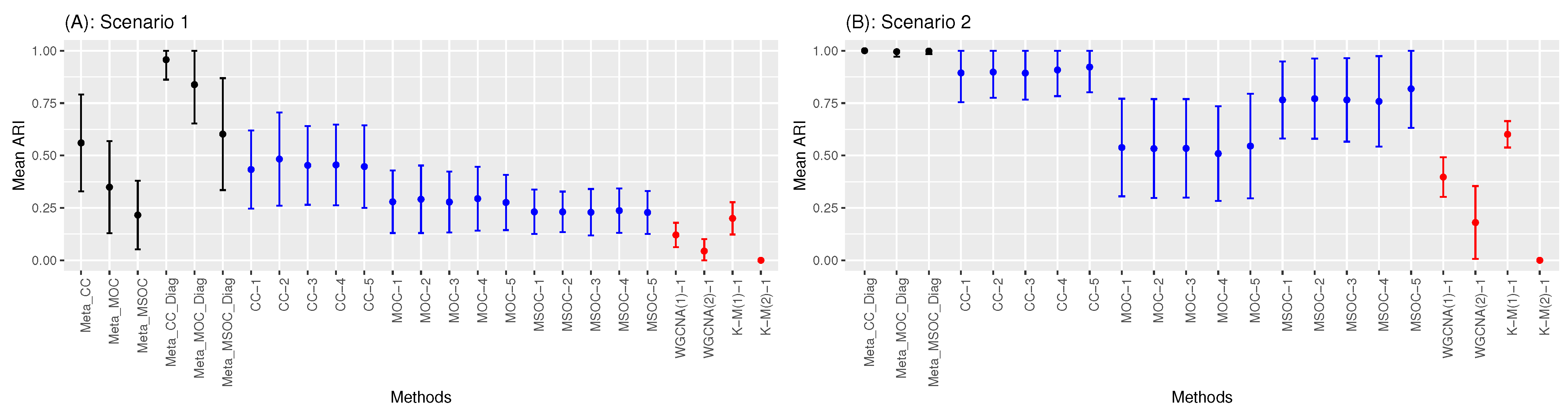
3.1. Scenario 1: The Within-Gene Correlation () Is Greater or Equal to the Correlation between the Same Type of Omics of Different Genes of the Same Cluster ()
3.1.1. Case 1: All The Studies Are Independent and Identically Distributed (iid)
3.1.2. Case 2: A Mixture of Two Groups of iid Studies with Different Values of p
3.1.3. Case 3: None of the Studies Are iid with Different Values of
3.2. Scenario 2: The Within-Gene Correlation () Is Less than or Equal to the Correlation between the Same Type of Omics of Different Genes of the Same Cluster ()
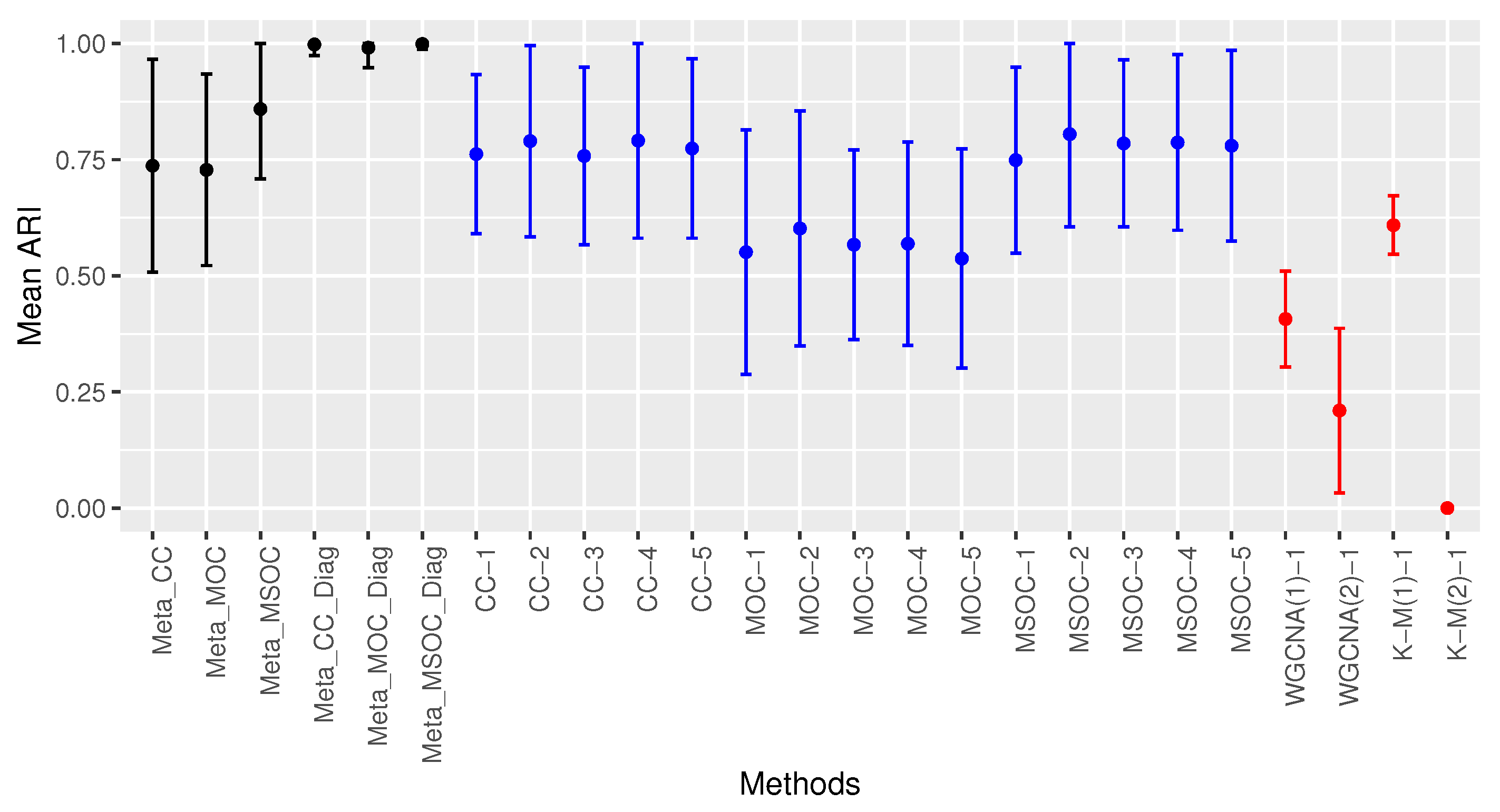
3.3. Low Signal with Small Sample Size
- The diagonal correlation matrix methods still outperform the whole matrix ones;
- The multi-study methods still improve the performance of the related single-study methods;
- The modified WGCNA methods still outperform the traditional one.
4. Real Data Analysis
- Removing the genes with missing values;
- Matching the ensemble gene ID version to the HUGO gene symbol in the RNA dataset;
- Taking the average of duplicated samples if the average correlation of duplicates is higher than the average correlation of non-duplicate samples; otherwise, randomly selecting one of the duplicates;
- Taking the gene symbol with the highest coefficient of variation among duplicated gene symbols;
- Matching the probe ID to the HUGO gene symbol in the methylation dataset using the Illumina annotation file "HM27.hg38.manifest.gencode.v36.tsv" (downloaded from https://zwdzwd.github.io/InfiniumAnnotation#human, accessed date 30 May 2024);
- Taking the first listed gene symbol when the probe ID was associated with several genes;
- Taking average of methylation beta values across all CpG sites within the same gene;
- Normalization by applying the variance stabilizing transformation (implemented by vst function in DESeq2 R package) to the RNA-seq count data;
- Keeping the samples that are common to both datasets and getting rid of the genes with zero expression level in all those samples.
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sethi, J.K.; Hotamisligil, G.S. Metabolic Messengers: Tumour necrosis factor. Nat. Metab. 2021, 3, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Heir, R.; Stellwagen, D. TNF-Mediated Homeostatic Synaptic Plasticity: From in vitro to in vivo Models. Front. Cell. Neurosci. 2020, 14, 565841. [Google Scholar] [CrossRef] [PubMed]
- Gough, P.; Myles, I.A. Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects. Front. Immunol. 2020, 11, 585880. [Google Scholar] [CrossRef] [PubMed]
- Oyelade, J.; Isewon, I.; Oladipupo, F.; Aromolaran, O.; Uwoghiren, E.; Ameh, F.; Achas, M.; Adebiyi, E. Clustering Algorithms: Their Application to Gene Expression Data. Bioinform. Biol. Insights 2016, 10, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Yoshikuni, Y. Pathway and Gene Discovery from Natural Hosts and Organisms. Methods Mol. Biol. 2019, 1927, 1–9. [Google Scholar] [CrossRef]
- Prasad, V.; Fojo, T.; Brada, M. Precision oncology: Origins, optimism, and potential. Lancet Oncol. 2016, 17, e81–e86. [Google Scholar] [CrossRef] [PubMed]
- Rappoport, N.; Shamir, R. Multi-omic and multi-view clustering algorithms: Re-view and cancer benchmark. Nucleic Acids Res. 2018, 46, 10546–10562. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Härdle, W.; Simar, L. Canonical Correlation Analysis. In Applied Multivariate Statistical Analysis; Springer: Berlin/Heidelberg, Germany, 2007; pp. 321–330. [Google Scholar] [CrossRef]
- Hafdahl, A.R. Combining Correlation Matrices: Simulation Analysis of Improved Fixed-Effects Methods. J. Educ. Behav. Stat. 2007, 32, 180–205. [Google Scholar] [CrossRef]
- Olkin, I.; Siotani, M. Asymptotic distribution of functions of a correlation matrix. In Essays in Provability and Statistics: A Volume in Honor of Professor Junjiro Ogawa; Shinko Tsusho: Tokyo, Japan, 1976; pp. 235–251. [Google Scholar]
- Muirhead, R.J.; Waternaux, C.M. Asymptotic distributions in canonical correlation analysis and other multivariate procedures for nonnormal populations. Biometrika 1980, 67, 31–43. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A General Framework for Weighted Gene Co-Expression Network Analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Yip, A.M.; Horvath, S. Gene network interconnectedness and the generalized topological overlap measure. BMC Bioinform. 2007, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Ravasz, E.; Somera, A.L.; Mongru, D.A.; Oltvai, Z.N.; Barabási, A.L. Hierarchical organization of modularity in metabolic networks. Science 2002, 297, 1551–1555. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, F. 8. Hierarchical Clustering. In Introduction to HPC with MPI for Data Science; Springer: Berlin/Heidelberg, Germany, 2016; pp. 32–58. ISBN 978-3-319-21903-5. [Google Scholar]
- Hartigan, J.A.; Wong, M.A. Algorithm AS 136: A k-Means Clustering Algorithm. J. R. Stat. Soc. C Appl. Stat. 1979, 28, 100–108. [Google Scholar] [CrossRef]
- Tibshirani, R.; Walther, G.; Hastie, T. Estimating the number of clusters in a dataset via the gap statistic. J. R. Stat. Soc. B Stat. Methodol. 2001, 63, 411–423. [Google Scholar] [CrossRef]
- Hubert, L.; Arabie, P. Comparing Partitions. J. Classif. 1985, 2, 193–218. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kemmo Tsafack, U.; Ahn, K.W.; Kwitek, A.E.; Lin, C.-W. Meta-Analytic Gene-Clustering Algorithm for Integrating Multi-Omics and Multi-Study Data. Bioengineering 2024, 11, 587. https://doi.org/10.3390/bioengineering11060587
Kemmo Tsafack U, Ahn KW, Kwitek AE, Lin C-W. Meta-Analytic Gene-Clustering Algorithm for Integrating Multi-Omics and Multi-Study Data. Bioengineering. 2024; 11(6):587. https://doi.org/10.3390/bioengineering11060587
Chicago/Turabian StyleKemmo Tsafack, Ulrich, Kwang Woo Ahn, Anne E. Kwitek, and Chien-Wei Lin. 2024. "Meta-Analytic Gene-Clustering Algorithm for Integrating Multi-Omics and Multi-Study Data" Bioengineering 11, no. 6: 587. https://doi.org/10.3390/bioengineering11060587
APA StyleKemmo Tsafack, U., Ahn, K. W., Kwitek, A. E., & Lin, C.-W. (2024). Meta-Analytic Gene-Clustering Algorithm for Integrating Multi-Omics and Multi-Study Data. Bioengineering, 11(6), 587. https://doi.org/10.3390/bioengineering11060587