

Leveraging Diverse Cell-Death Patterns to Decipher the Interactive Relation of Unfavorable Outcome and Tumor Microenvironment in Breast Cancer

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. The Single-Sample Gene Set Enrichment Analysis (ssGSEA) Algorithm
2.3. Tumor Microenvironment (TME) and Immunocyte Infiltration Analysis
2.4. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.5. Enrichment Analysis for Biological Function and Pathway
2.6. Identification of Differential Gene Expression (DEGs)
2.7. Survival Analysis
2.8. Gene Mutation Landscapes
2.9. Least Absolute Shrinkage and Selection Operator (LASSO)
2.10. Analysis of Affected Tumor-Related Pathways and Therapeutic Targets
2.11. Statistical Analysis
3. Results
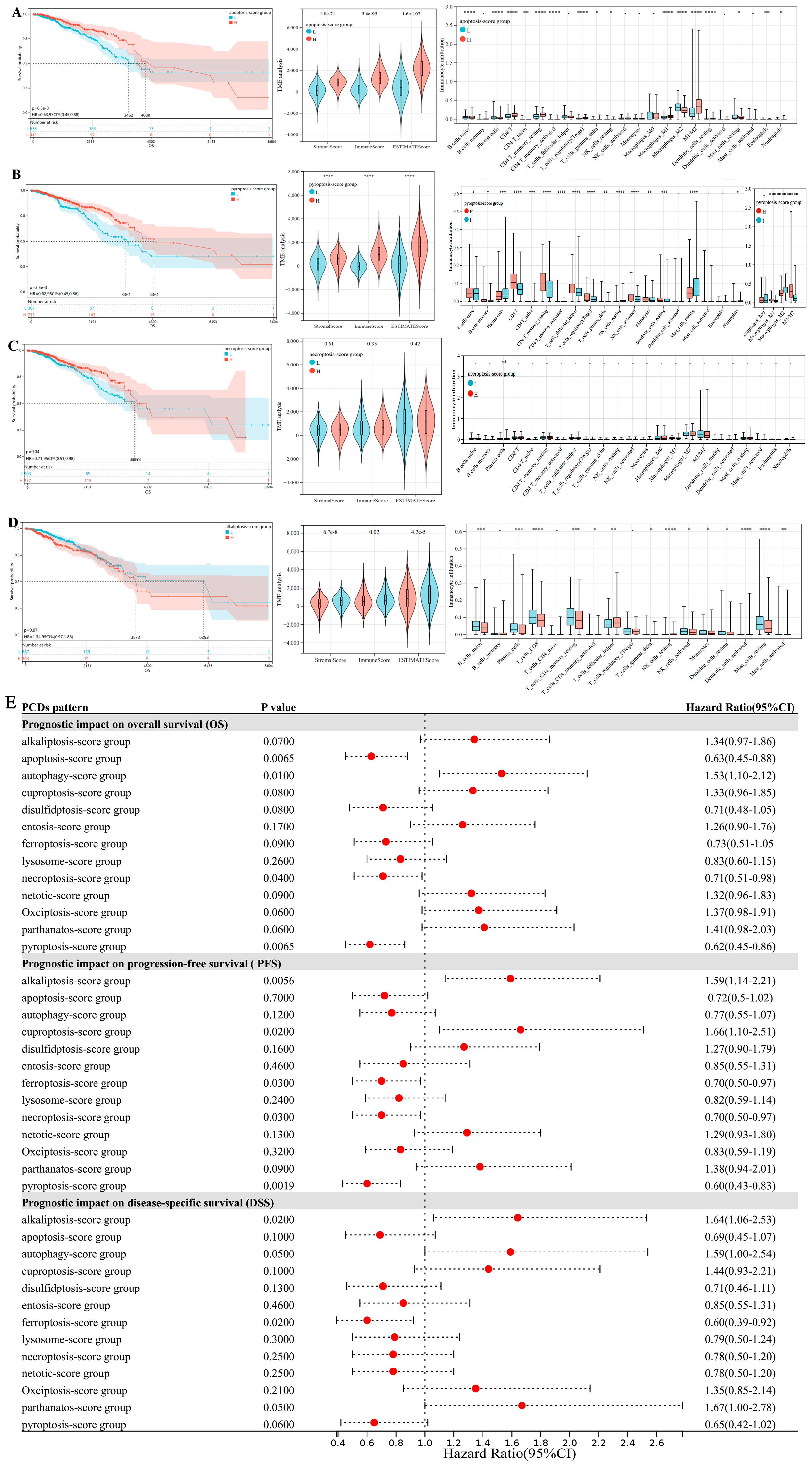
3.1. Prognostic Difference in Breast Cancer Affected by Expression States of 13 PCD Patterns
3.2. Co-Expression Networks Link PCD Gene Modules to Clinical Features in Breast Cancer
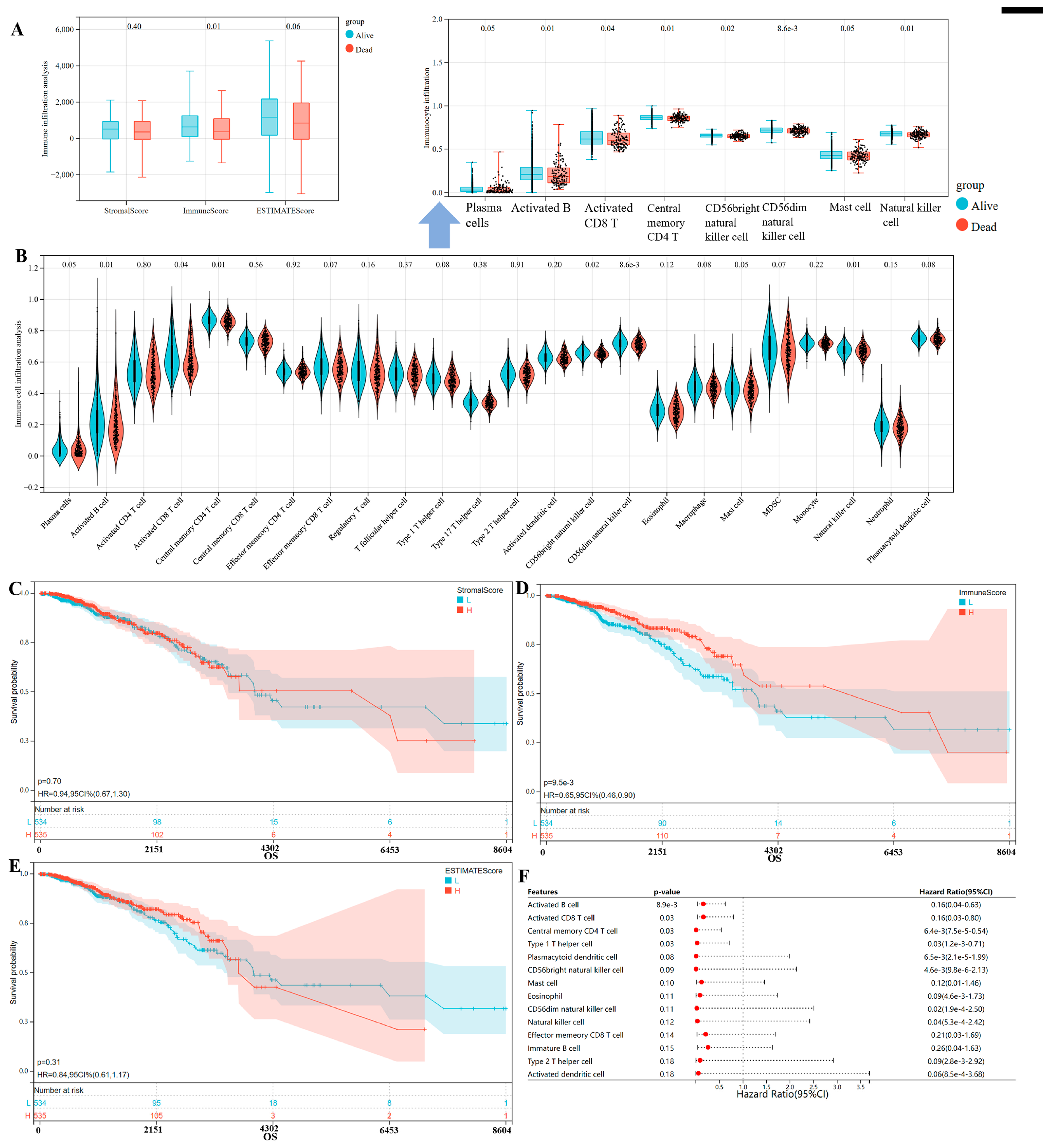
3.3. Immune Microenvironment Patterns with Different Survival Outcomes in Breast Cancer
3.4. The Correlations of Cell-Death Patterns (Apoptosis, Pyroptosis) and Immune Checkpoint Gene Expression
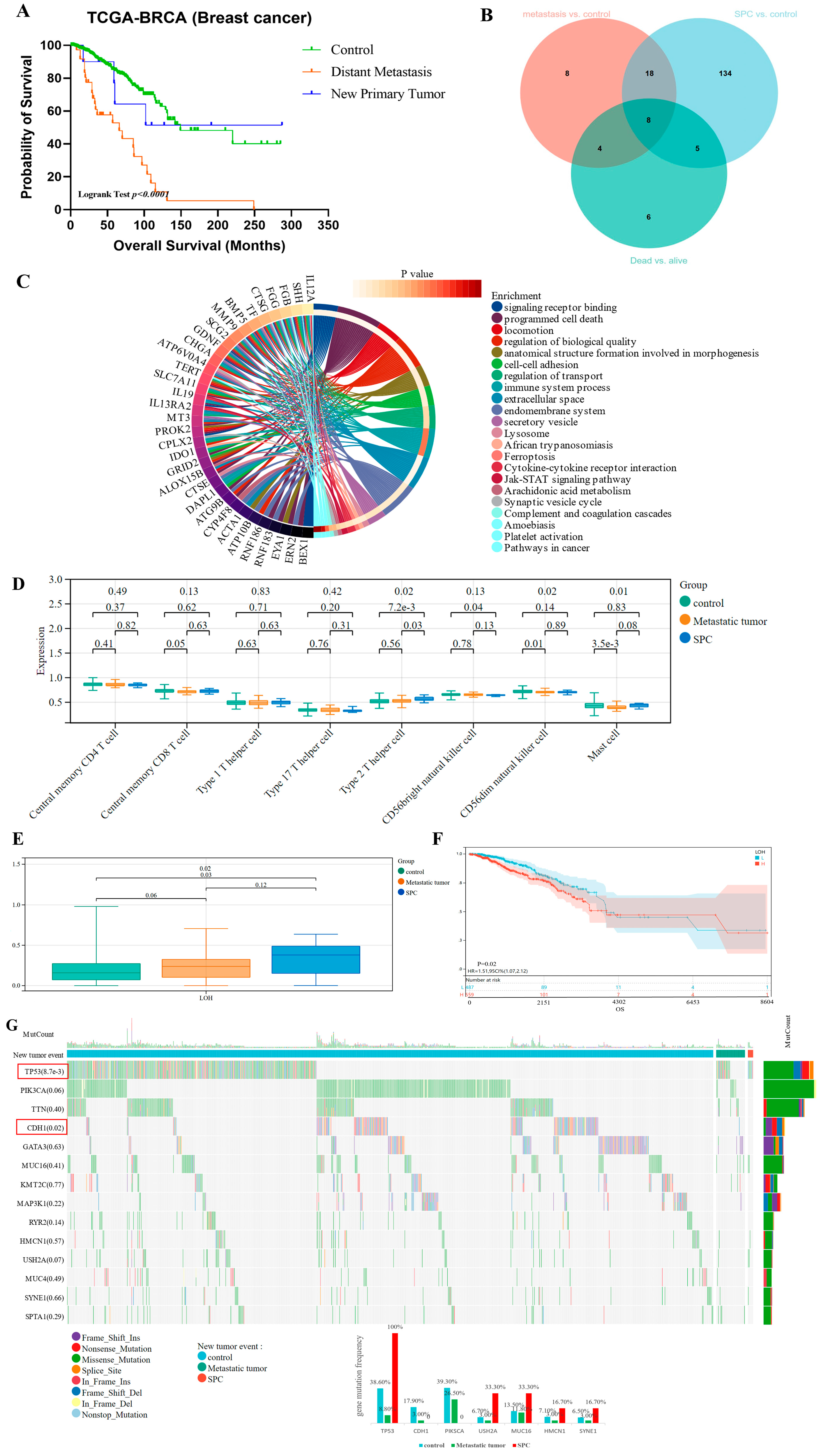
3.5. New Tumor Occurrence Caused Poor Prognosis of Breast Cancer Patients
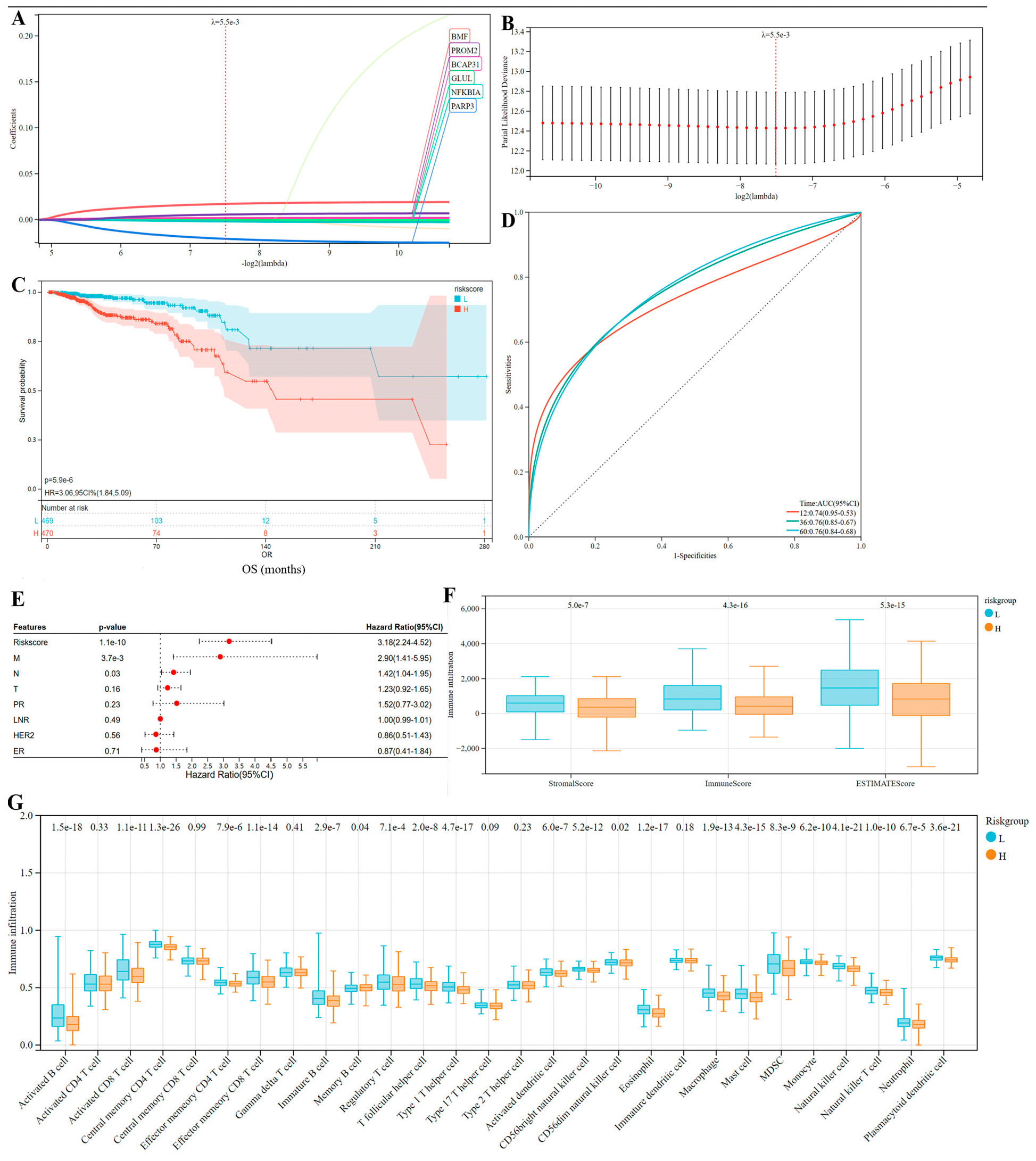
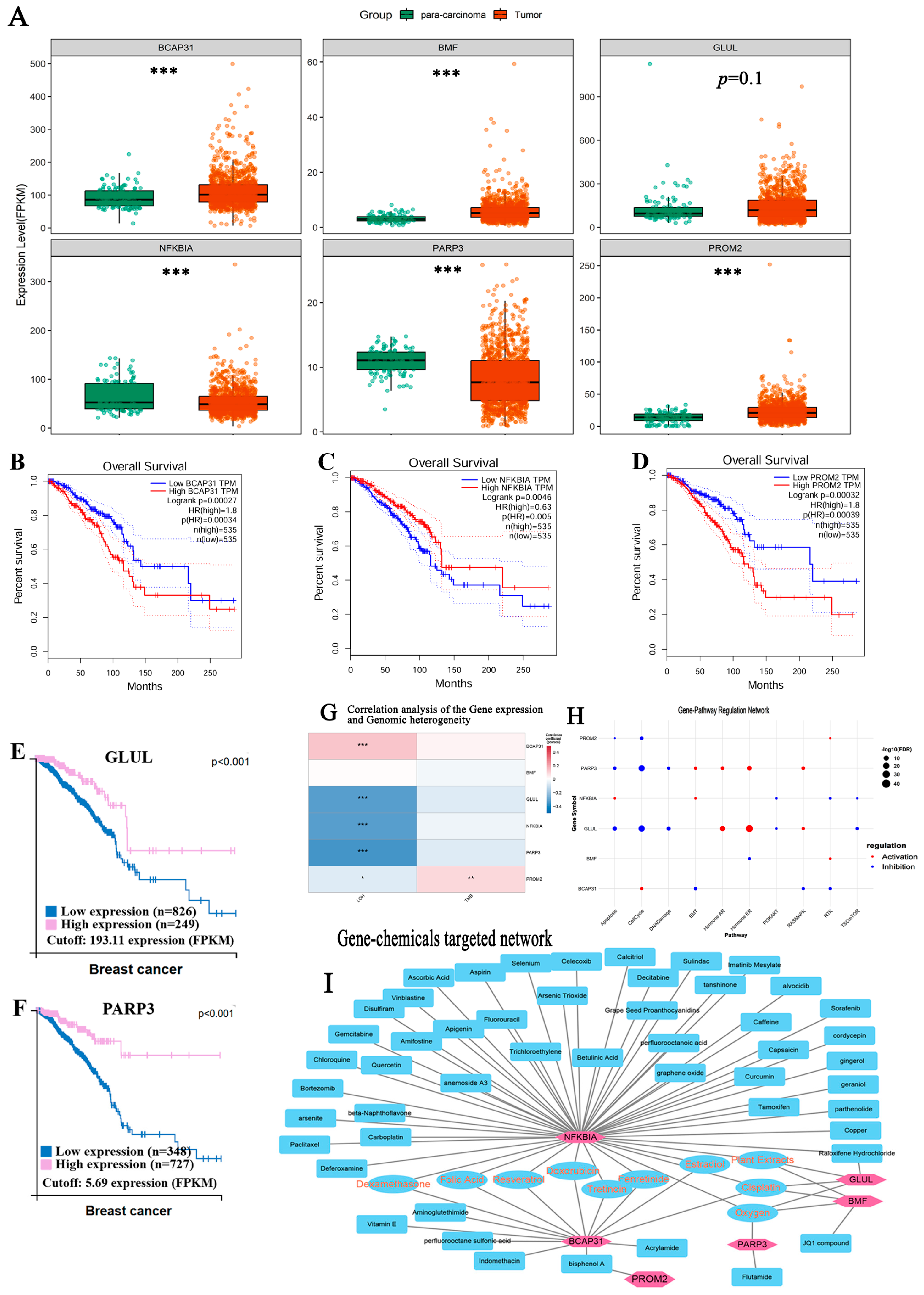
3.6. Identification of Markers Predicting the Prognosis of Breast Cancer
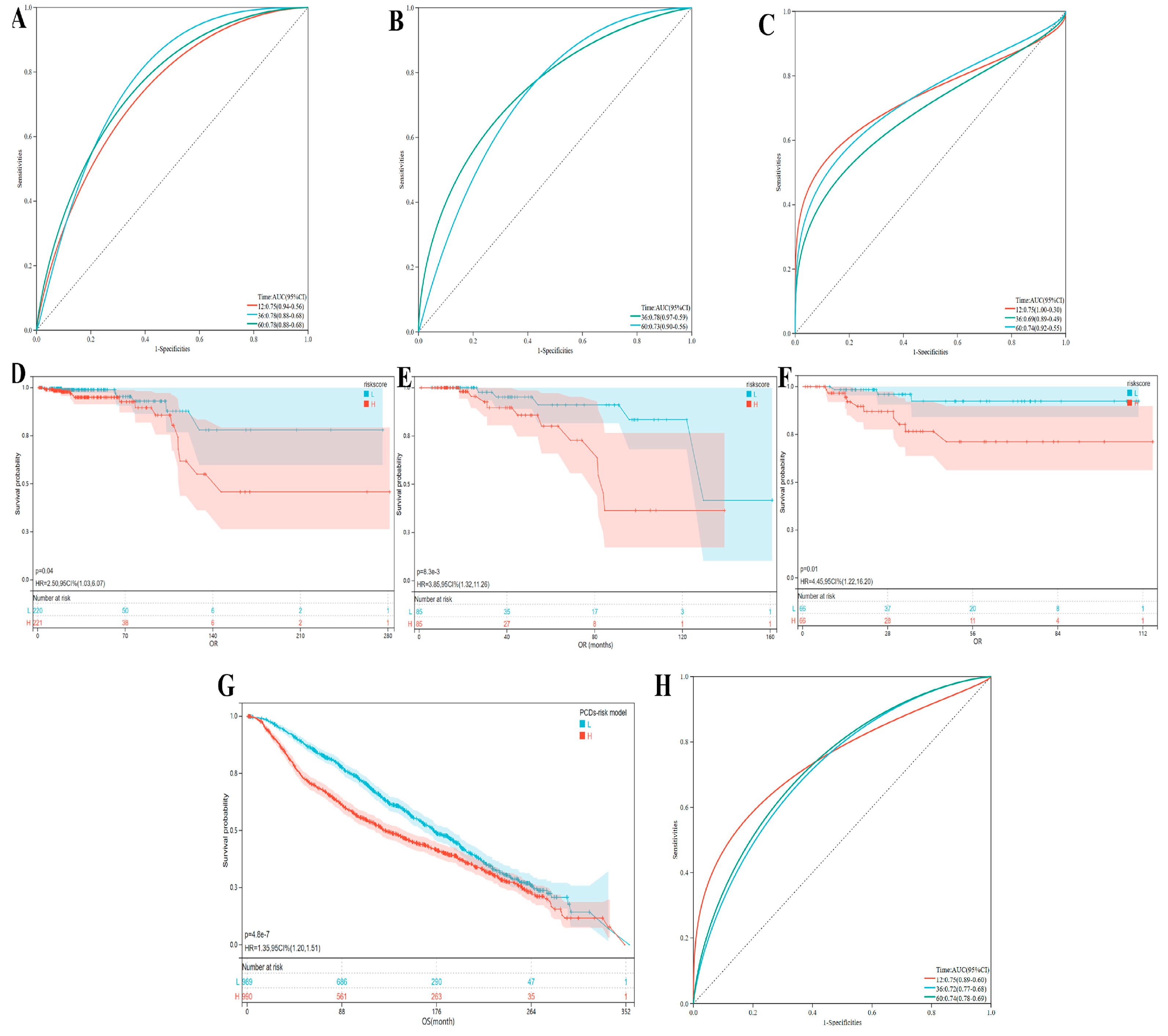
3.7. The Evaluation and Understanding of Model Performance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PCD | Programmed cell death |
| BC | Breast cancer |
| TME | Tumor microenvironment |
| LASSO | Least Absolute Shrinkage and Selection Operator |
| WGCNA | weighted gene co-expression network analysis |
| TCGA | The Cancer Genome Atlas |
| ssGSEA | single-sample Gene Set Enrichment Analysis |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| SPC | Second primary cancers |
| METABRIC | Molecular Taxonomy of Breast Cancer International Consortium |
| DEGs | Differential gene expression |
| TNBC | Triple-negative breast cancer |
References
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Haffty, B.G.; Yang, Q.; Reiss, M.; Kearney, T.; Higgins, S.A.; Weidhaas, J.; Harris, L.; Hait, W.; Toppmeyer, D. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 5652–5657. [Google Scholar] [CrossRef] [PubMed]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Liu, J.; Hong, M.; Li, Y.; Chen, D.; Wu, Y.; Hu, Y. Programmed Cell Death Tunes Tumor Immunity. Front. Immunol. 2022, 13, 847345. [Google Scholar] [CrossRef]
- Ershaid, N.; Sharon, Y.; Doron, H.; Raz, Y.; Shani, O.; Cohen, N.; Monteran, L.; Leider-Trejo, L.; Ben-Shmuel, A.; Yassin, M.; et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat. Commun. 2019, 10, 4375. [Google Scholar] [CrossRef]
- Wu, L.; Lu, H.; Pan, Y.; Liu, C.; Wang, J.; Chen, B.; Wang, Y. The role of pyroptosis and its crosstalk with immune therapy in breast cancer. Front. Immunol. 2022, 13, 973935. [Google Scholar] [CrossRef]
- Hage, C.; Hoves, S.; Strauss, L.; Bissinger, S.; Prinz, Y.; Pöschinger, T.; Kiessling, F.; Ries, C.H. Sorafenib Induces Pyroptosis in Macrophages and Triggers Natural Killer Cell-Mediated Cytotoxicity Against Hepatocellular Carcinoma. Hepatology 2019, 70, 1280–1297. [Google Scholar] [CrossRef]
- Zhu, S.; Zhao, Q.; Fan, Y.; Tang, C. Development of a prognostic model to predict BLCA based on anoikis-related gene signature: Preliminary findings. BMC Urol. 2023, 23, 199. [Google Scholar] [CrossRef]
- Liu, Q.; Liao, L. Identification of macrophage-related molecular subgroups and risk signature in colorectal cancer based on a bioinformatics analysis. Autoimmunity 2024, 57, 2321908. [Google Scholar] [CrossRef]
- Liu, C.-J.; Hu, F.-F.; Xie, G.-Y.; Miao, Y.-R.; Li, X.-W.; Zeng, Y.; Guo, A.-Y. GSCA: An integrated platform for gene set cancer analysis at genomic, pharmacogenomic and immunogenomic levels. Brief. Bioinform. 2023, 24, bbac558. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-J.; Hu, F.-F.; Xia, M.-X.; Han, L.; Zhang, Q.; Guo, A.-Y. GSCALite: A web server for gene set cancer analysis. Bioinformatics 2018, 34, 3771–3772. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yin, Y.; Zhang, T.; Wang, J.; Guo, Z.; Li, Y.; Zhao, Y.; Qin, R.; He, Q. A comprehensive landscape analysis of autophagy in cancer development and drug resistance. Front. Immunol. 2024, 15, 1412781. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rew. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautès–Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rew. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Jiang, Z.-B.; Huang, J.-M.; Xie, Y.-J.; Zhang, Y.Z.; Chang, C.; Lai, H.-L.; Wang, W.; Yao, X.-J.; Fan, X.-X.; Wu, Q.-B.; et al. Evodiamine suppresses non-small cell lung cancer by elevating CD8(+) T cells and downregulating the MUC1-C/PD-L1 axis. J. Exp. Clin. Cancer Res. CR 2020, 39, 249. [Google Scholar] [CrossRef]
- Chen, S.; Saeed, A.F.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef]
- Ghimirey, N.; Steele, C.; Czerniecki, B.J.; Koski, G.K.; Showalter, L.E. Sunitinib Combined with Th1 Cytokines Potentiates Apoptosis in Human Breast Cancer Cells and Suppresses Tumor Growth in a Murine Model of HER-2(pos) Breast Cancer. Int. J. Breast Cancer 2021, 2021, 8818393. [Google Scholar] [CrossRef]
- Crotty, S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014, 41, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Zhou, X.; Zheng, M.; Yu, Y.; Jin, G.; Zhang, S. Regulatory T cells are associated with the tumor immune microenvironment and immunotherapy response in triple-negative breast cancer. Front. Immunol. 2023, 14, 1263537. [Google Scholar] [CrossRef]
- Brantley, K.D.; Rosenberg, S.M.; Collins, L.C.; Ruddy, K.J.; Tamimi, R.M.; Schapira, L.; Borges, V.F.; Warner, E.; Come, S.E.; Zheng, Y.; et al. Second Primary Breast Cancer in Young Breast Cancer Survivors. JAMA Oncol. 2024, 10, 718–725. [Google Scholar] [CrossRef]
- Zhang, N.-N.; Sun, Q.-S.; Chen, Z.; Liu, F.; Jiang, Y.-Y. Homeostatic regulatory role of Pokemon in NF-κB signaling: Stimulating both p65 and IκBα expression in human hepatocellular carcinoma cells. Mol. Cell. Biochem. 2013, 372, 57–64. [Google Scholar] [CrossRef]
- Badr, E.A.; Assar, M.F.; Eltorgoman, A.M.A.; Labeeb, A.Z.; Breaka, G.A.; Elkhouly, E.A. A correlation between BCL-2 modifying factor, p53 and livin gene expressions in cancer colon patients. Biochem. Biophys. Rep. 2020, 22, 100747. [Google Scholar] [CrossRef]
- Xu, F.; Xia, T.; Xu, Q.-T.; Zhang, X.; Huang, Y.-Z.; Sun, X.; Shi, L.; Zhou, X.-J.; Wei, J.-F.; Ding, Q. RBMS2 Chemosensitizes Breast Cancer Cells to Doxorubicin by Regulating BMF Expression. Int. J. Biol. Sci. 2022, 18, 1724–1736. [Google Scholar] [CrossRef]
- Bott, A.J.; Shen, J.; Tonelli, C.; Zhan, L.; Sivaram, N.; Jiang, Y.-P.; Yu, X.; Bhatt, V.; Chiles, E.; Zhong, H.; et al. Glutamine Anabolism Plays a Critical Role in Pancreatic Cancer by Coupling Carbon and Nitrogen Metabolism. Cell Rep. 2019, 29, 1287–1298.e6. [Google Scholar] [CrossRef]
- Lim, Y.X.; Lin, H.; Chu, T.; Lim, Y.P. WBP2 promotes BTRC mRNA stability to drive migration and invasion in triple-negative breast cancer via NF-κB activation. Mol. Oncol. 2022, 16, 422–446. [Google Scholar] [CrossRef]
- Zuo, H.; Yang, D.; Yang, Q.; Tang, H.; Fu, Y.-X.; Wan, Y. Differential regulation of breast cancer bone metastasis by PARP1 and PARP2. Nat. Commun. 2020, 11, 1578. [Google Scholar] [CrossRef]
- Wang, L.; Wang, D.; Sonzogni, O.; Ke, S.; Wang, Q.; Thavamani, A.; Batalini, F.; Stopka, S.A.; Regan, M.S.; Vandal, S.; et al. PARP-inhibition reprograms macrophages toward an anti-tumor phenotype. Cell Rep. 2022, 41, 111462. [Google Scholar] [CrossRef] [PubMed]
- Morganti, S.; Marra, A.; De Angelis, C.; Toss, A.; Licata, L.; Giugliano, F.; Salimbeni, B.T.; Giachetti, P.P.M.B.; Esposito, A.; Giordano, A.; et al. PARP Inhibitors for Breast Cancer Treatment: A Review. JAMA Oncol. 2024, 10, 658–670. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Shi, J.; Yang, C.; Luo, J.; Li, Y.; Ren, J.; Lv, Y.; Zhang, Y. Regulation of tumorigenesis and ferroptosis in non-small cell lung cancer by a novel BBOX1-AS1/miR-326/PROM2 axis. Mol. Cell. Biochem. 2024, 479, 2143–2155. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distant Metastasis (Metastatic Tumor) | New Primary Tumor | Control (Follow up Without New Neoplasm Occurrence) | p-Value | |
|---|---|---|---|---|
| Age | 57.3 ± 12.5 | 50.9 ± 12.0 | 58.7 ± 13.2 | 0.15 |
| Survival status | Dead 69.4% (25/36) | Dead 40.0% (4/10) | Dead 11.3% (115/1020) | 1.4e-24 |
| T stage | 0.75 | |||
| T1 | 19.4% (7/36) | 40% (4/10) | 26.2% (267/1020) | |
| T2 | 55.6% (20/36) | 60% (6/10) | 58.1% (593/1020) | |
| T3 | 19.4% (7/36) | 0 | 12.4% (126/1020) | |
| T4 | 5.6% (2/36) | 0 | 3.3% (34/1020) | |
| N stage | 0.03 | |||
| N0 | 22.2% (8/36) | 60% (6/10) | 49.9% (509/1020) | |
| N1 | 44.4% (16/36) | 30% (3/10) | 32.4% (331/1020) | |
| N2 | 16.7% (6/36) | 10% (1/10) | 11.0% (112/1020) | |
| N3 | 16.7% (6/36) | 0 | 6.7% (68/1020) | |
| M stage | 0.02 | |||
| M0 | 91.7% (33/36) | 100% | 98.1% (1001/1020) | |
| M1 | 8.3% (3/36) | 0 | 1.9% (19/1020) | |
| Stage | 7.9e-7 | |||
| Stage I | 5.9% (2/34) | 22.2% (2/9) | 17.5% (179/1020) | |
| Stage II | 41.2% (14/34) | 66.7% (6/9) | 58.6% (598/1020) | |
| Stage III | 44.1% (15/34) | 11.1% (1/9) | 22.5% (230/1020) | |
| Stage IV | 8.8% (3/34) | 0 | 1.9% (19/1020) | |
| Immune phenotype | ||||
| ER− | 35.3% (12/34) | 60.0% (6/10) | 24.0% (216/990) | 0.05 |
| PR− | 58.8% (20/34) | 70.0% (7/10) | 31.7% (313/987) | 6.5e-3 |
| HER2 | 0.03 | |||
| amplifications | 18.2% (4/22) | 0 | 19.8% (181/913) | |
| HER2− | 81.8% (18/22) | 100.0% (6/6) | 80.2% (732/913) | |
| TNBC | 22.7% (5/22) | 66.7% (4/6) | 24.5% (224/913) | 0.05 |
| Lymph nodes | 0.04 | |||
| positive tests | 76.3% (29/38) | 44.4% (4/9) | 49.8% (435/873) | |
| LNR | 24.9 ± 29.1 | 6.7 ± 11.7 | 16.6 ± 27.0 | |
| Margin status+ | 13.9% (5/36) | 0 | 7.6% (72/945) | 0.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Ding, T.; Zhang, T.; Liu, S.; Wang, J.; Zhou, X.; Guo, Z.; He, Q.; Zhang, S. Leveraging Diverse Cell-Death Patterns to Decipher the Interactive Relation of Unfavorable Outcome and Tumor Microenvironment in Breast Cancer. Bioengineering 2025, 12, 420. https://doi.org/10.3390/bioengineering12040420
Li Y, Ding T, Zhang T, Liu S, Wang J, Zhou X, Guo Z, He Q, Zhang S. Leveraging Diverse Cell-Death Patterns to Decipher the Interactive Relation of Unfavorable Outcome and Tumor Microenvironment in Breast Cancer. Bioengineering. 2025; 12(4):420. https://doi.org/10.3390/bioengineering12040420
Chicago/Turabian StyleLi, Yue, Ting Ding, Tong Zhang, Shuangyu Liu, Jinhua Wang, Xiaoyan Zhou, Zeqi Guo, Qian He, and Shuqun Zhang. 2025. "Leveraging Diverse Cell-Death Patterns to Decipher the Interactive Relation of Unfavorable Outcome and Tumor Microenvironment in Breast Cancer" Bioengineering 12, no. 4: 420. https://doi.org/10.3390/bioengineering12040420
APA StyleLi, Y., Ding, T., Zhang, T., Liu, S., Wang, J., Zhou, X., Guo, Z., He, Q., & Zhang, S. (2025). Leveraging Diverse Cell-Death Patterns to Decipher the Interactive Relation of Unfavorable Outcome and Tumor Microenvironment in Breast Cancer. Bioengineering, 12(4), 420. https://doi.org/10.3390/bioengineering12040420