Microfluidic Protein Imaging Platform: Study of Tau Protein Aggregation and Alzheimer’s Drug Response

Abstract
:1. Introduction
2. Materials and Methods
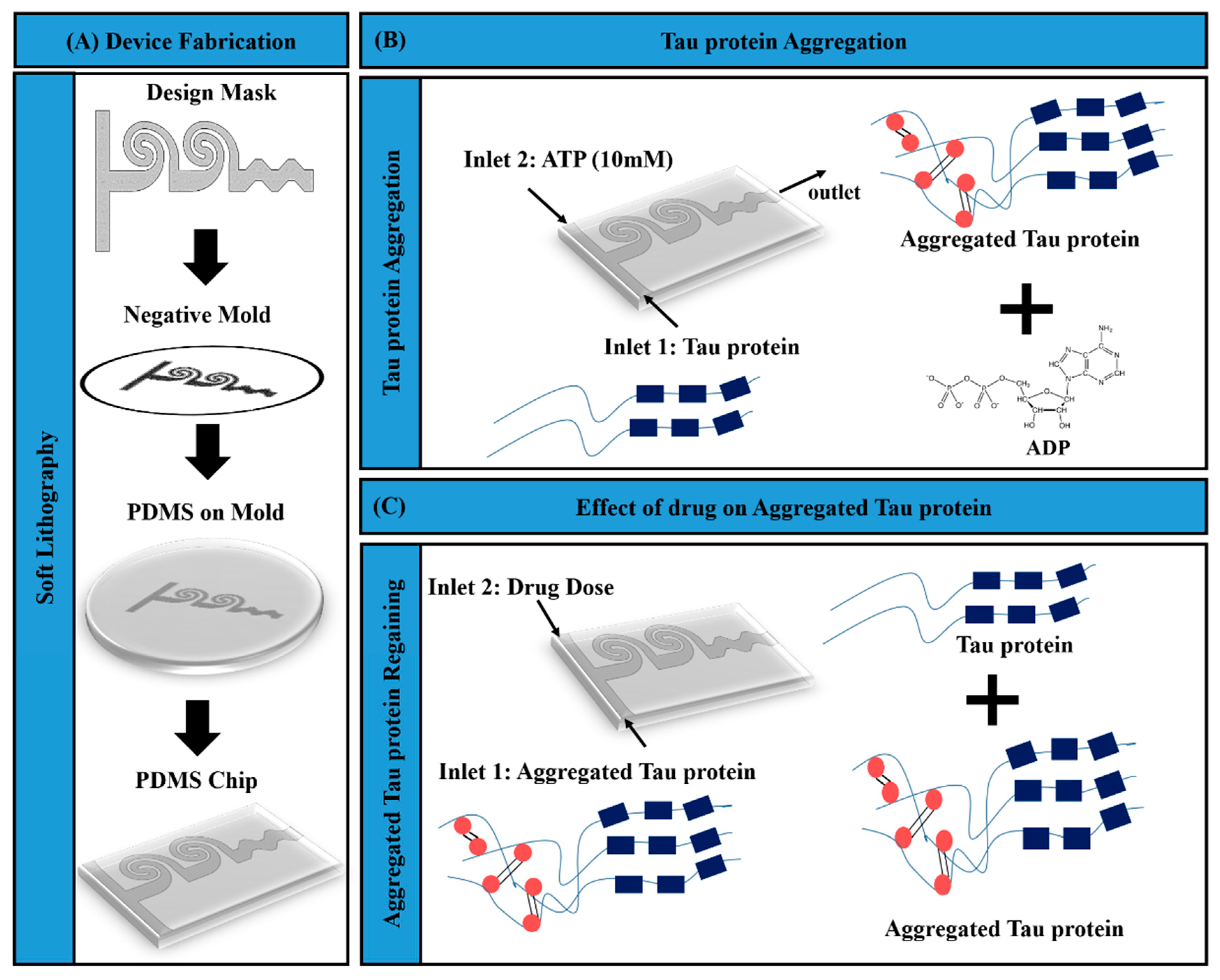
2.1. Schematic of Microfluidic Assay for Tau Aggregation and Drug Response
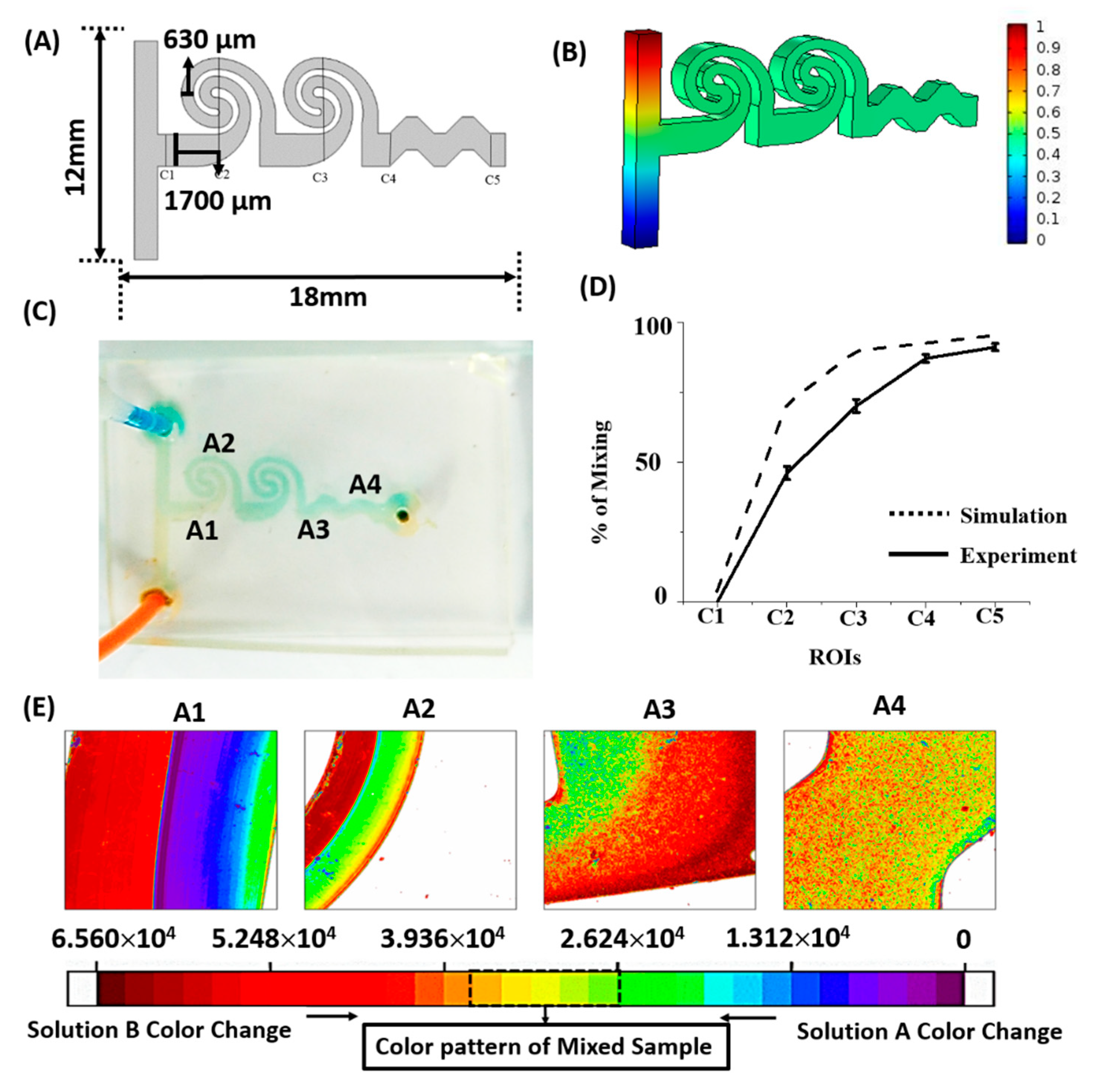
2.2. Fabrication of Microfluidic Mixer
2.3. Modeling and Validation of Microfluidic Mixer Design
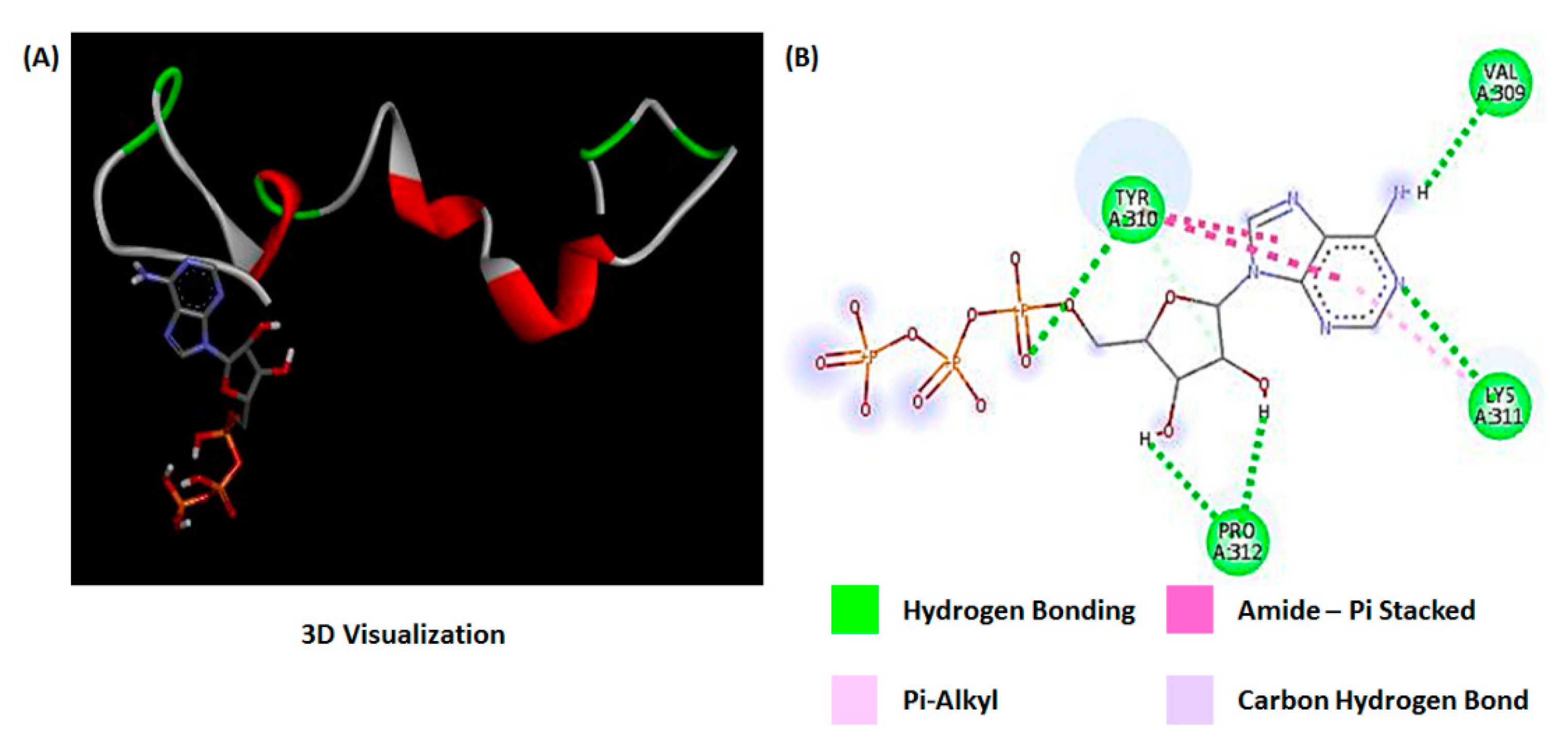
2.4. Molecular Modeling of Tau Protein Aggregation
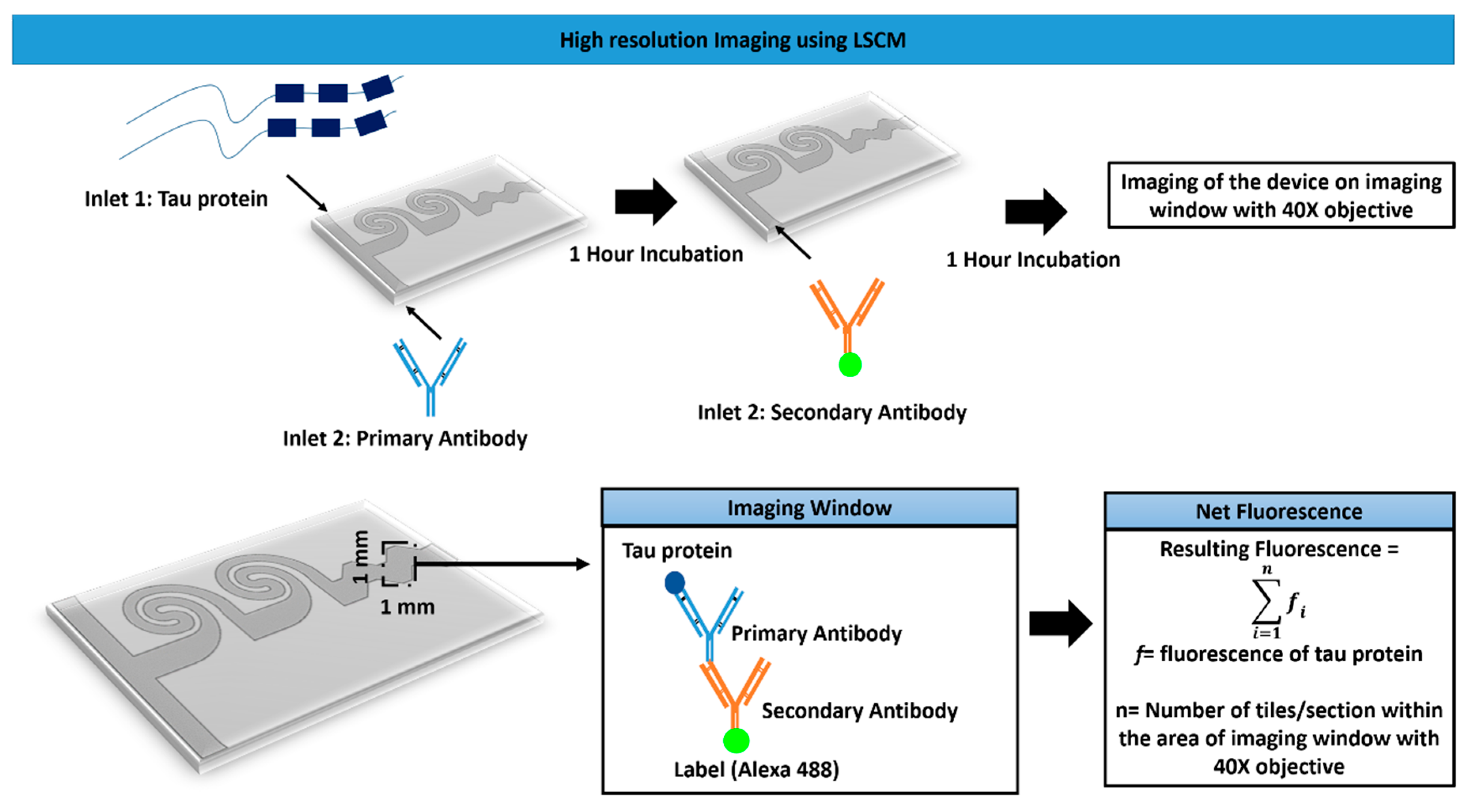
2.5. Microfluidic Experiments for Tau Imaging
- f = fluorescence of tau protein;
- n = number of tiles/sections within the area of the imaging window.
3. Results and Discussion
3.1. Micromixing Simulations
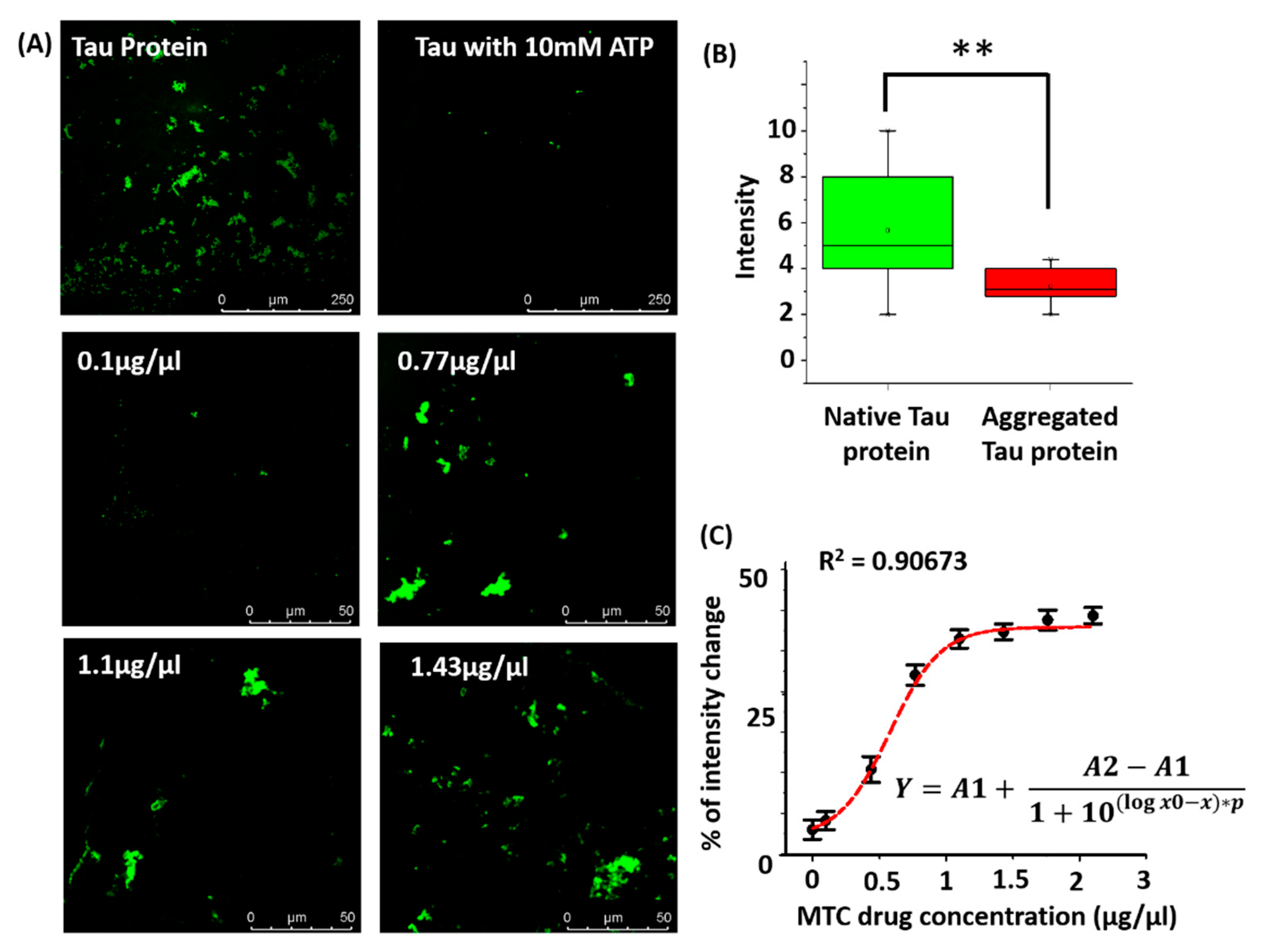
3.2. Tau Aggregation—Molecular Simulation and Experiments
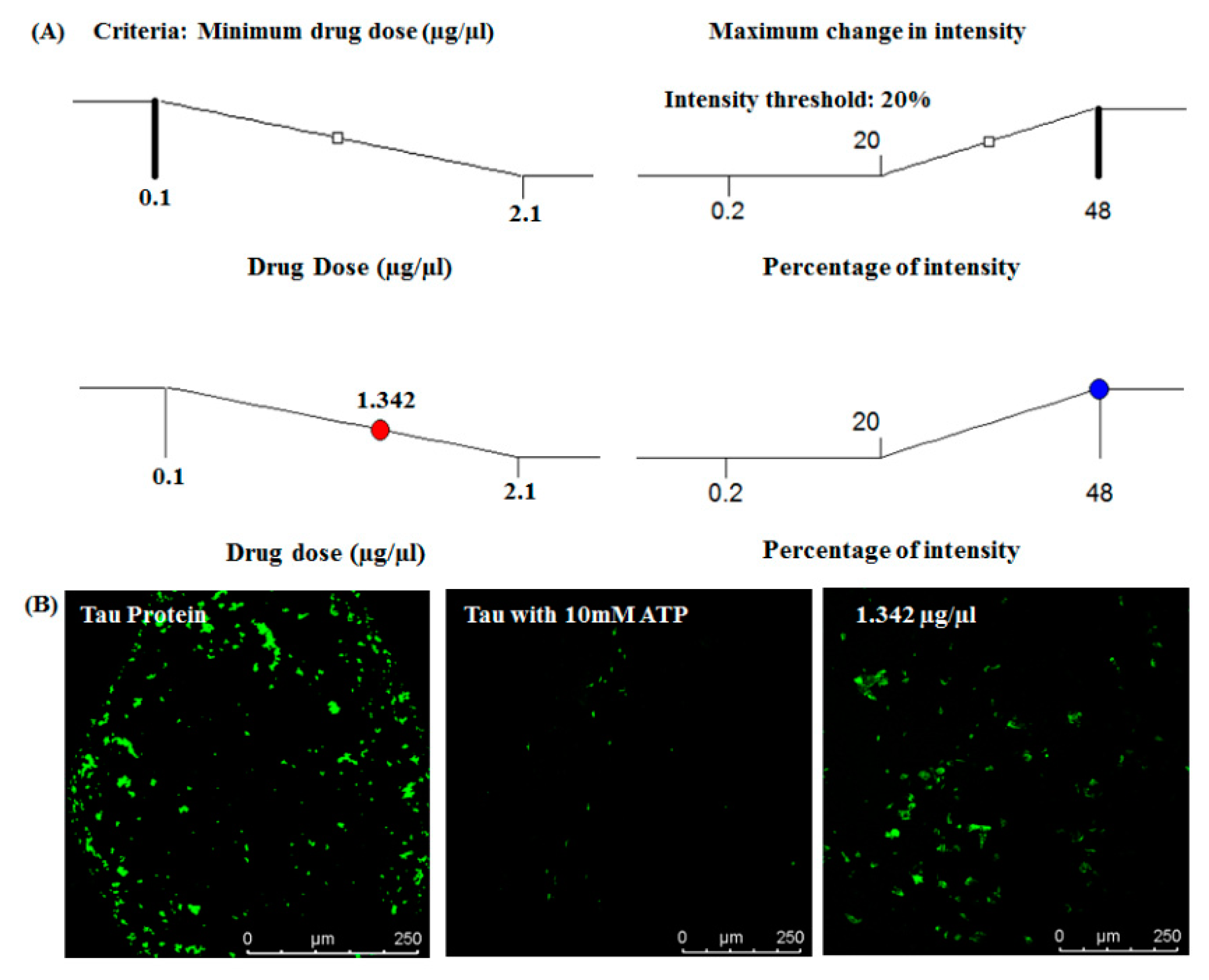
3.3. Drug Dose Optimization
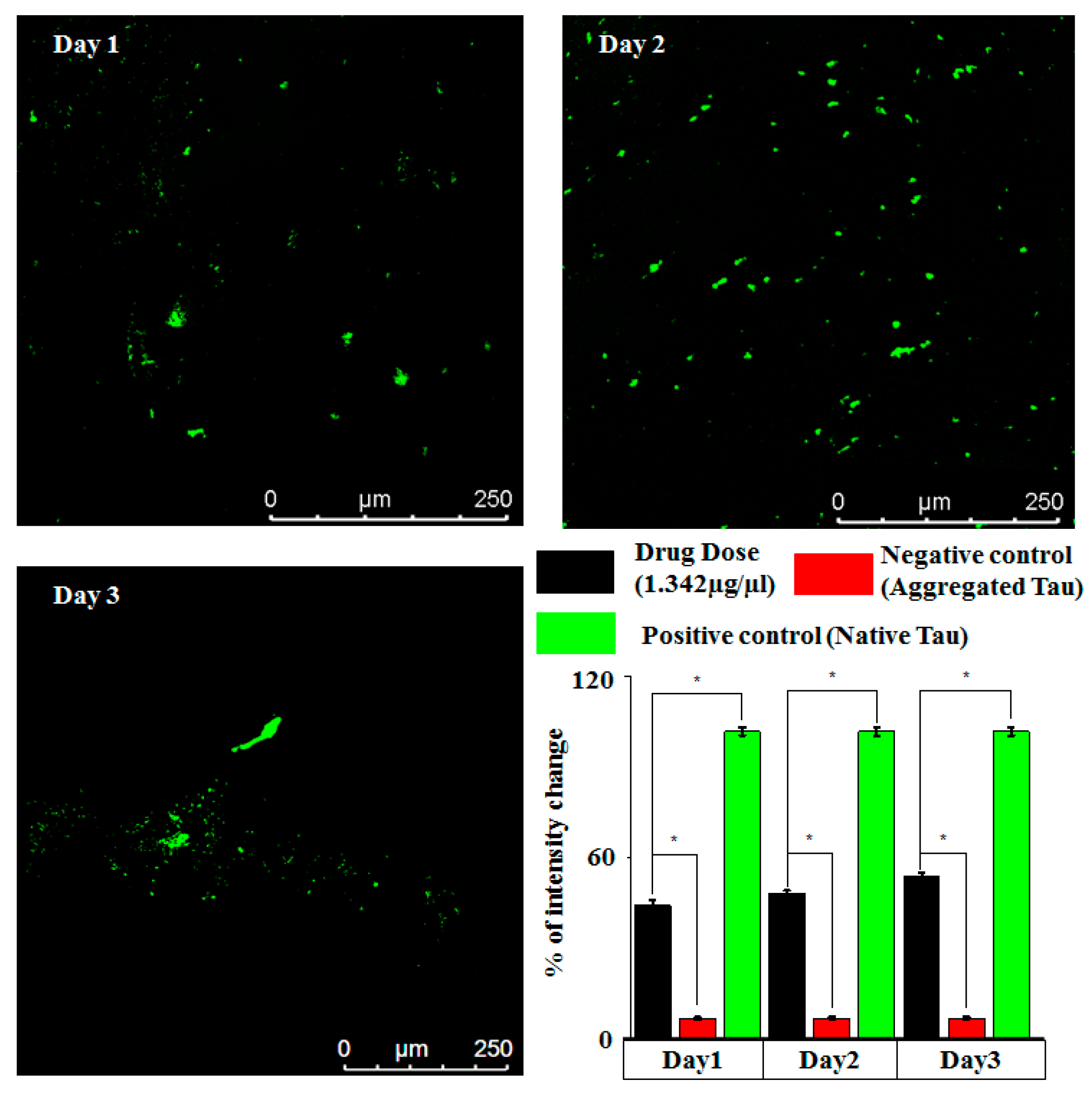
3.4. On-Chip Tau Aggregation and Neurological Implications
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gong, C.-X.; Grundke-Iqbal, I.; Iqbal, K. Targeting Tau Protein in Alzheimer’s Disease. Drugs Aging 2010, 27, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Revisiting atherosclerosis and dementia. Nat. Neurosci. 2020, 23, 691–692. [Google Scholar] [CrossRef] [PubMed]
- Penney, J.; Ralvenius, W.T.; Tsai, L.-H. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol. Psychiatry 2020, 25, 148–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Small, S.A.; Gandy, S. Sorting through the Cell Biology of Alzheimer’s Disease: Intracellular Pathways to Pathogenesis. Neuron 2006, 52, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Ballatore, C.; Lee, V.M.-Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef]
- Irwin, D.J.; Lee, V.M.-Y.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative Tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Farid, M.; Corbo, C.P.; Alonso, A.D. Tau binds ATP and induces its aggregation. Microsc. Res. Tech. 2013, 77, 133–137. [Google Scholar] [CrossRef]
- Shammas, S.L.; Garcia, G.A.; Kumar, S.; Kjaergaard, M.; Horrocks, M.H.; Shivji, N.; Mandelkow, E.; Knowles, T.P.; Mandelkow, E.; Klenerman, D. A mechanistic model of tau amyloid aggregation based on direct observation of oligomers. Nat. Commun. 2015, 6, 7025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wischik, C.M.; Harrington, C.R.; Storey, J.M. Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 529–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, T.P.; Levin, A.; Knowles, T.P.; Dobson, C.M.; Knowles, T.P. Microfluidic approaches for probing amyloid assembly and behaviour. Lab Chip 2018, 18, 999–1016. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, C.; Feng, X.; Xu, Y.; Liu, B.-F. Ultrafast microfluidic mixer for tracking the early folding kinetics of human telomere G-quadruplex. Anal. Chem. 2014, 86, 4333–4339. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, Y.; Feng, X.; Liu, B.-F. A rapid microfluidic mixer for high-viscosity fluids to track ultrafast early folding kinetics of g-quadruplex under molecular crowding conditions. Anal. Chem. 2012, 84, 9025–9032. [Google Scholar] [CrossRef] [PubMed]
- Perry, S.L.; Guha, S.; Pawate, A.S.; Bhaskarla, A.; Agarwal, V.; Nair, S.K.; Kenis, P.J.A. A microfluidic approach for protein structure determination at room temperature via on-chip anomalous diffraction. Lab Chip 2013, 13, 3183–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldauer, S.A.; Wu, L.; Yu, M.; Bakajin, O.; Lapidus, L.J. Microfluidic mixers for studying protein folding. J. Vis. Exp. 2012, e3976. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Zeng, Y.; Sun, Q.; Sun, Y.; Guo, Z.; Qu, J.Y.; Yao, S. Microsecond Protein Folding Events Revealed by Time-Resolved Fluorescence Resonance Energy Transfer in a Microfluidic Mixer. Anal. Chem. 2015, 87, 5589–5595. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Li, Y.; Chen, P.; Feng, X.; Du, W.; Liu, B.-F. Rapid three-dimensional microfluidic mixer for high viscosity solutions to unravel earlier folding kinetics of G-quadruplex under molecular crowding conditions. Talanta 2016, 149, 237–243. [Google Scholar] [CrossRef]
- Qin, L.; Vermesh, O.; Shi, Q.; Heath, J.R. Self-powered microfluidic chips for multiplexed protein assays from whole blood. Lab Chip 2009, 9, 2016–2020. [Google Scholar] [CrossRef] [Green Version]
- Ghonge, T.; Koydemir, H.C.; Valera, E.; Berger, J.; Garcia, C.; Nawar, N.; Tiao, J.; Damhorst, G.; Ganguli, A.; Hassan, U.; et al. Smartphone-imaged microfluidic biochip for measuring CD64 expression from whole blood. Analyst 2019, 144, 3925–3935. [Google Scholar] [CrossRef] [PubMed]
- Kilic, O.; Pamies, D.; Lavell, E.; Schiapparelli, P.; Feng, Y.; Hartung, T.; Bal-Price, A.; Hogberg, H.T.; Quiñones-Hinojosa, A.; Guerrero-Cazares, H.; et al. Brain-on-a-chip model enables analysis of human neuronal differentiation and chemotaxis. Lab Chip 2016, 16, 4152–4162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, E.L.; Hachi, S.; Hemmer, K.; Trietsch, S.J.; Baumuratov, A.S.; Hankemeier, T.; Vulto, P.; Schwamborn, J.C.; Fleming, R.M.T. Differentiation of neuroepithelial stem cells into functional dopaminergic neurons in 3D microfluidic cell culture. Lab Chip 2015, 15, 2419–2428. [Google Scholar] [CrossRef] [PubMed]
- Haring, A.P.; Sontheimer, H.; Johnson, B.N. Microphysiological Human Brain and Neural Systems-on-a-Chip: Potential Alternatives to Small Animal Models and Emerging Platforms for Drug Discovery and Personalized Medicine. Stem Cell Rev. Rep. 2017, 13, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Sudarsan, A.P.; Ugaz, V.M. Fluid mixing in planar spiral microchannels. Lab Chip 2006, 6, 74–82. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Tetz, G.; Pinho, M.; Pritzkow, S.; Mendez, N.; Soto, C.; Tetz, V. Bacterial DNA promotes Tau aggregation. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.; Roth, M.; Harrington, C.R. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. USA 1996, 93, 11213–11218. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, R.P.; Tepper, K.; Rönicke, R.; Soom, M.; Westermann, M.; Reymann, K.; Kaether, C.; Fändrich, M. Mechanism of amyloid plaque formation suggests an intracellular basis of Aβ pathogenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 1942–1947. [Google Scholar] [CrossRef] [Green Version]
- Koenigsknecht, J. Microglial Phagocytosis of fibrillar β-amyloid through a β1 integrin-dependent mechanism. J. Neurosci. 2004, 24, 9838–9846. [Google Scholar] [CrossRef] [Green Version]
- Panza, F.; Solfrizzi, V.; Seripa, D.; Imbimbo, B.P.; Lozupone, M.; Santamato, A.; Zecca, C.; Barulli, M.R.; Bellomo, A.; Pilotto, A.; et al. Tau-Centric Targets and Drugs in Clinical Development for the Treatment of Alzheimer’s Disease. BioMed Res. Int. 2016, 2016, 3245935. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Blurton-Jones, M.; Rhee, S.W.; Cribbs, D.H.; Cotman, C.W.; Jeon, N.L. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat. Methods 2005, 2, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Wallin, A.; Ågren, H.; Spenger, C.; Siegfried, J.; Vanmechelen, E. Tau protein in cerebrospinal fluid. Mol. Chem. Neuropathol. 1995, 26, 231–245. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | Factor 1 (Drug Dose µg/µL) | Response 1 (% of Intensity) |
|---|---|---|
| 1 | 0.1 | 0.2 |
| 2 | 1.43 | 48 |
| 3 | 1.76 | 47.5 |
| 4 | 0.77 | 28.4 |
| 5 | 2.1 | 47.89 |
| 6 | 1.1 | 45.23 |
| 7 | 2.1 | 47.44 |
| 8 | 1.1 | 46.02 |
| 9 | 0.44 | 1.6 |
| 10 | 0.1 | 0.31 |
| Protein Target | Ligand | Ligand Efficiency (kcal/mol) | Inhibition Constant | Electrostatic Energy | Electrostatic Interaction | Pi-Pi Interaction |
|---|---|---|---|---|---|---|
| Tau | ATP | −0.02 | 319.75 | −0.41 | NA | TYR310 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jain, S.; Swain, S.; Das, L.; Swain, S.; Giri, L.; Kondapi, A.K.; Narayanan Unni, H. Microfluidic Protein Imaging Platform: Study of Tau Protein Aggregation and Alzheimer’s Drug Response. Bioengineering 2020, 7, 162. https://doi.org/10.3390/bioengineering7040162
Jain S, Swain S, Das L, Swain S, Giri L, Kondapi AK, Narayanan Unni H. Microfluidic Protein Imaging Platform: Study of Tau Protein Aggregation and Alzheimer’s Drug Response. Bioengineering. 2020; 7(4):162. https://doi.org/10.3390/bioengineering7040162
Chicago/Turabian StyleJain, Shubha, Sarpras Swain, Lopamudra Das, Sarita Swain, Lopamudra Giri, Anand Kumar Kondapi, and Harikrishnan Narayanan Unni. 2020. "Microfluidic Protein Imaging Platform: Study of Tau Protein Aggregation and Alzheimer’s Drug Response" Bioengineering 7, no. 4: 162. https://doi.org/10.3390/bioengineering7040162
APA StyleJain, S., Swain, S., Das, L., Swain, S., Giri, L., Kondapi, A. K., & Narayanan Unni, H. (2020). Microfluidic Protein Imaging Platform: Study of Tau Protein Aggregation and Alzheimer’s Drug Response. Bioengineering, 7(4), 162. https://doi.org/10.3390/bioengineering7040162