
Bone Marrow Multipotent Mesenchymal Stromal Cells as Autologous Therapy for Osteonecrosis: Effects of Age and Underlying Causes





Abstract
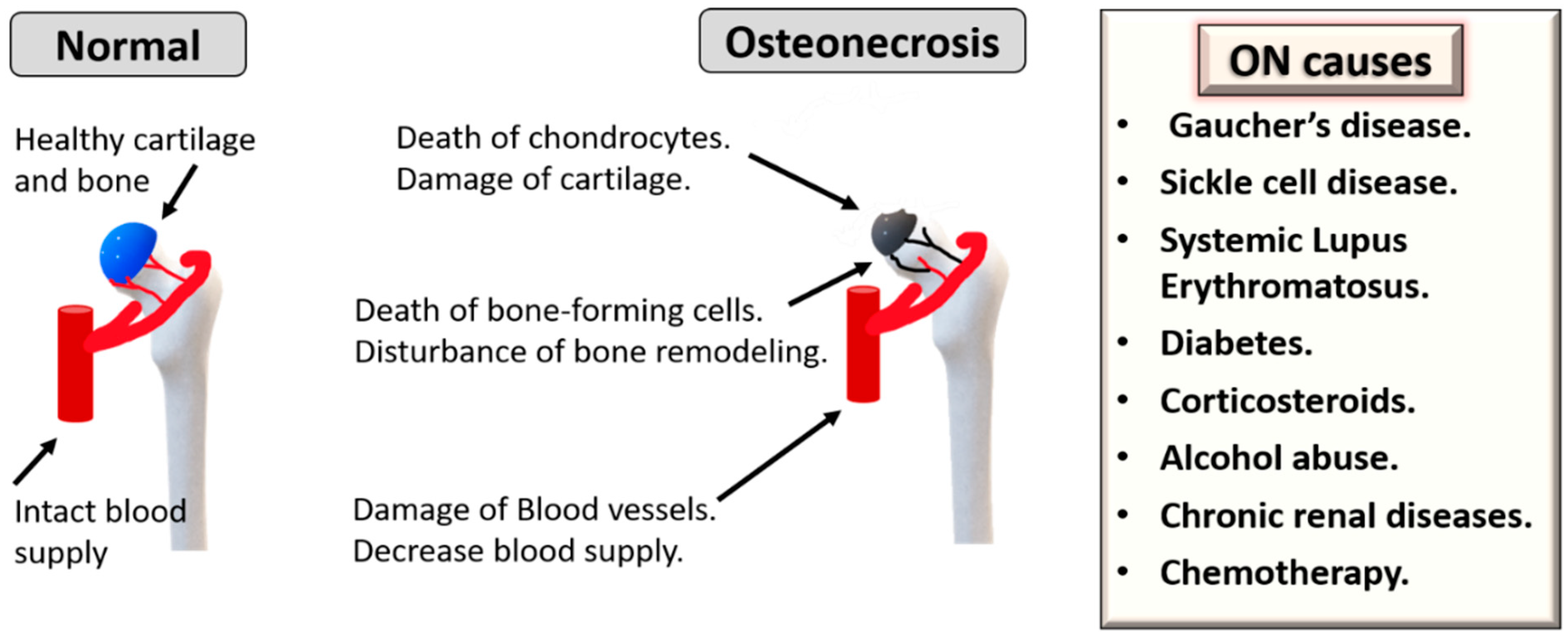
:1. Introduction
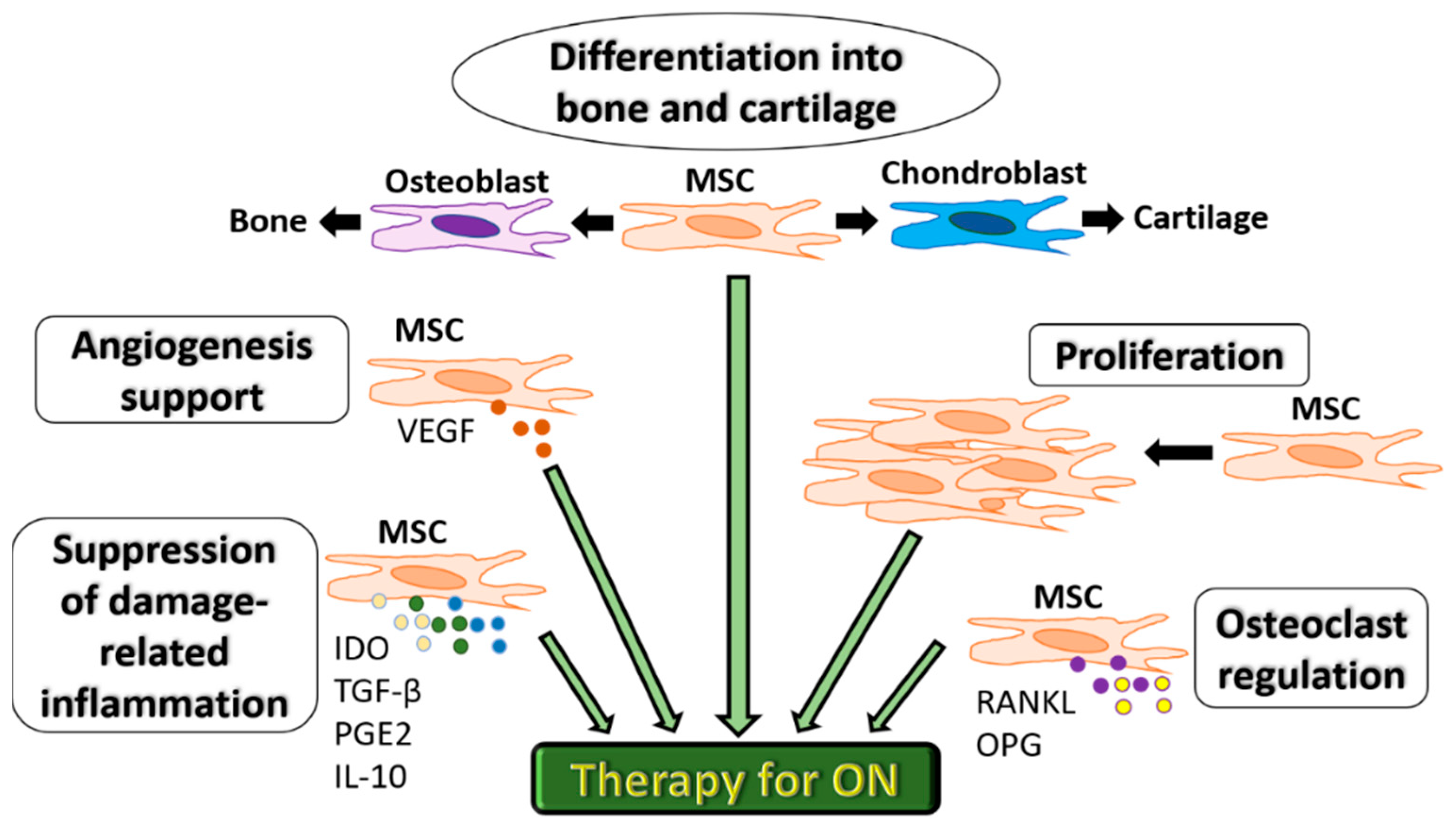
2. MSC Therapy for Osteonecrosis
2.1. Pre-Clinical Studies
2.2. Clinical Studies
3. Age-Related Changes in MSCs
3.1. Pre-Clinical Studies
3.2. Clinical/Human Studies
4. The Effect of Glucocorticoids on MSCs
4.1. Pre-Clinical Studies
4.2. Clinical/Human Studies
5. Alcohol Effects on MSCs
5.1. Pre-Clinical Studies
5.2. Clinical/Human Studies
6. MSCs in Sickle Cell Disease
6.1. Pre-Clinical Studies
6.2. Clinical/Human Studies
7. MSCs in Gaucher Disease
7.1. Pre-Clinical Studies
7.2. Clinical/Human Studies
8. MSCs in Systemic Lupus Erythematosus
8.1. Pre-Clinical Studies
8.2. Clinical/Human Studies
9. MSCs in Diabetes
9.1. Pre-Clinical Studies
9.2. Clinical/Human Studies
10. Chronic Kidney Diseases Effects on MSCs
10.1. Pre-Clinical Studies
10.2. Clinical/Human Studies
11. Chemotherapy Effects on MSCs
11.1. Pre-Clinical Studies
11.2. Clinical/Human Studies
12. Conclusions: Challenges and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Shen, S.; Fu, H.; Wang, Z.; Li, X.; Sui, X.; Yuan, M.; Liu, S.; Wang, G.; Guo, Q. Immunomodulatory Functions of Mesenchymal Stem Cells in Tissue Engineering. Stem Cells Int. 2019, 20199671206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernigou, P.; Poignard, A.; Beaujean, F.; Rouard, H. Percutaneous autologous bone-marrow grafting for nonunions. Influence of the number and concentration of progenitor cells. J. Bone Jt. Surg. Am. 2005, 87, 1430–1437. [Google Scholar] [CrossRef]
- Katagiri, W.; Kawai, T.; Osugi, M.; Sugimura-Wakayama, Y.; Sakaguchi, K.; Kojima, T.; Kobayashi, T. Angiogenesis in newly regenerated bone by secretomes of human mesenchymal stem cells. Maxillofac. Plast. Reconstr. Surg. 2017, 39, 8. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.A.; Kinsey, S.E.; English, A.; Jones, R.A.; Straszynski, L.; Meredith, D.M.; Markham, A.F.; Jack, A.; Emery, P.; McGonagle, D. Isolation and characterization of bone marrow multipotential mesenchymal progenitor cells. Arthritis Rheum. 2002, 46, 3349–3360. [Google Scholar] [CrossRef] [PubMed]
- Elgaz, S.; Bonig, H.; Bader, P. Mesenchymal stromal cells for osteonecrosis. J. Transl. Med. 2020, 18, 399. [Google Scholar] [CrossRef]
- Hernigou, P.; Trousselier, M.; Roubineau, F.; Bouthors, C.; Chevallier, N.; Rouard, H.; Flouzat-Lachaniette, C.H. Stem Cell Therapy for the Treatment of Hip Osteonecrosis: A 30-Year Review of Progress. Clin. Orthop. Surg. 2016, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Shigemura, T.; Nakamura, J.; Kishida, S.; Harada, Y.; Ohtori, S.; Kamikawa, K.; Ochiai, N.; Takahashi, K. Incidence of osteonecrosis associated with corticosteroid therapy among different underlying diseases: Prospective MRI study. Rheumatology 2011, 50, 2023–2028. [Google Scholar] [CrossRef] [Green Version]
- Dima, A.; Pedersen, A.B.; Pedersen, L.; Baicus, C.; Thomsen, R.W. Association of common comorbidities with osteonecrosis: A nationwide population-based case-control study in Denmark. BMJ Open 2018, 8, e020680. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Olsen, B.R. The roles of vascular endothelial growth factor in bone repair and regeneration. Bone 2016, 9130–9138. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Sumi, K.; Kunimatsu, R.; Oki, N.; Tsuka, Y.; Nakajima, K.; Ando, K.; Tanimoto, K. The effect of mesenchymal stem cells on osteoclast precursor cell differentiation. J. Oral. Sci. 2019, 61, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- El-Jawhari, J.J.; Jones, E.; Giannoudis, P.V. The roles of immune cells in bone healing; what we know, do not know and future perspectives. Injury 2016, 47, 2399–2406. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.B.; Maruyama, M. Inflammation, Bone Healing and Osteonecrosis: From Bedside to Bench. J. Inflamm. Res. 2020, 13913–13923. [Google Scholar] [CrossRef]
- Weiss, A.R.R.; Dahlke, M.H. Immunomodulation by Mesenchymal Stem Cells (MSCs): Mechanisms of Action of Living, Apoptotic, and Dead MSCs. Front. Immunol. 2019, 101191. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Cui, Y.; Zhang, M.; Zhao, D.; Liu, G.; Ding, J. Engineered three-dimensional scaffolds for enhanced bone regeneration in osteonecrosis. Bioact. Mater. 2020, 5, 584–601. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, G.; Johnson, B.N.; Jia, X. Three-dimensional (3D) printed scaffold and material selection for bone repair. Acta Biomater. 2019, 8416–8433. [Google Scholar] [CrossRef]
- Maruyama, M.; Nabeshima, A.; Pan, C.C.; Behn, A.W.; Thio, T.; Lin, T.; Pajarinen, J.; Kawai, T.; Takagi, M.; Goodman, S.B.; et al. The effects of a functionally-graded scaffold and bone marrow-derived mononuclear cells on steroid-induced femoral head osteonecrosis. Biomaterials 2018, 18739–18746. [Google Scholar] [CrossRef]
- Peng, J.; Wen, C.; Wang, A.; Wang, Y.; Xu, W.; Zhao, B.; Zhang, L.; Lu, S.; Qin, L.; Guo, Q.; et al. Micro-CT-based bone ceramic scaffolding and its performance after seeding with mesenchymal stem cells for repair of load-bearing bone defect in canine femoral head. J. Biomed. Mater. Res. B Appl. Biomater. 2011, 96, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yang, F.; Wei, X.; Zhang, X.; Zhang, Y.; Wang, B.; Liu, G.; Xie, H.; Yang, J.; Wang, W.; et al. An exploratory study of articular cartilage and subchondral bone reconstruction with bone marrow mesenchymal stem cells combined with porous tantalum/Bio-Gide collagen membrane in osteonecrosis of the femoral head. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 99, 1123–1132. [Google Scholar] [CrossRef]
- Fan, L.; Zhang, C.; Yu, Z.; Shi, Z.; Dang, X.; Wang, K. Transplantation of hypoxia preconditioned bone marrow mesenchymal stem cells enhances angiogenesis and osteogenesis in rabbit femoral head osteonecrosis. Bone 2015, 81544–81553. [Google Scholar] [CrossRef]
- Ismail, T.; Osinga, R.; Todorov, A.; Haumer, A., Jr.; Tchang, L.A.; Epple, C.; Allafi, N.; Menzi, N.; Largo, R.D.; Kaempfen, A.; et al. Engineered, axially-vascularized osteogenic grafts from human adipose-derived cells to treat avascular necrosis of bone in a rat model. Acta Biomater. 2017, 63236–63245. [Google Scholar] [CrossRef] [PubMed]
- Phipps, M.C.; Monte, F.; Mehta, M.; Kim, H.K. Intraosseous Delivery of Bone Morphogenic Protein-2 Using a Self-Assembling Peptide Hydrogel. Biomacromolecules 2016, 17, 2329–2336. [Google Scholar] [CrossRef]
- Wang, C.K.; Ho, M.L.; Wang, G.J.; Chang, J.K.; Chen, C.H.; Fu, Y.C.; Fu, H.H. Controlled-release of rhBMP-2 carriers in the regeneration of osteonecrotic bone. Biomaterials 2009, 30, 4178–4186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.X.; Zhang, X.P.; Xiao, G.Y.; Hou, Y.; Cheng, L.; Si, M.; Wang, S.S.; Li, Y.H.; Nie, L. In vitro and in vivo evaluation of calcium phosphate composite scaffolds containing BMP-VEGF loaded PLGA microspheres for the treatment of avascular necrosis of the femoral head. Mater. Sci. Eng. C. Mater. Biol. Appl. 2016, 60298–60307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Wang, Y.M.; Chu, K.; Wang, Z.H.; Liu, Y.H.; Jiang, L.H.; Chen, X.; Zhou, Z.Y.; Yin, G. The application of PRP combined with TCP in repairing avascular necrosis of the femoral head after femoral neck fracture in rabbit. Eur. Rev. Med. Pharm. Sci. 2018, 22, 903–909. [Google Scholar] [CrossRef]
- Altaie, A.; Baboolal, T.G.; Wall, O.; Jones, E.; McGonagle, D. Platelet lysate enhances synovial fluid multipotential stromal cells functions: Implications for therapeutic use. Cytotherapy 2018, 20, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Vun, J.; Panteli, M.; Jones, E.; Giannoudis, P.V. The in vitro effects of platelet products on the biophysiological functions of human bone marrow mesenchymal stromal cells: A systematic review. Eur. Cell. Mater. 2021, 41269–41315. [Google Scholar] [CrossRef]
- Pak, J.; Lee, J.H.; Jeon, J.H.; Lee, S.H. Complete resolution of avascular necrosis of the human femoral head treated with adipose tissue-derived stem cells and platelet-rich plasma. J. Int. Med. Res. 2014, 42, 1353–1362. [Google Scholar] [CrossRef]
- Zhang, F.; Peng, W.X.; Wang, L.; Zhang, J.; Dong, W.T.; Wu, J.H.; Zhang, H.; Wang, J.B.; Zhao, Y. Role of FGF-2 Transfected Bone Marrow Mesenchymal Stem Cells in Engineered Bone Tissue for Repair of Avascular Necrosis of Femoral Head in Rabbits. Cell. Physiol. Biochem. 2018, 48, 773–784. [Google Scholar] [CrossRef]
- Peng, W.X.; Wang, L. Adenovirus-Mediated Expression of BMP-2 and BFGF in Bone Marrow Mesenchymal Stem Cells Combined with Demineralized Bone Matrix For Repair of Femoral Head Osteonecrosis in Beagle Dogs. Cell. Physiol. Biochem. 2017, 43, 1648–1662. [Google Scholar] [CrossRef] [PubMed]
- Muller, I.; Vaegler, M.; Holzwarth, C.; Tzaribatchev, N.; Pfister, S.M.; Schutt, B.; Reize, P.; Greil, J.; Handgretinger, R.; Rudert, M. Secretion of angiogenic proteins by human multipotent mesenchymal stromal cells and their clinical potential in the treatment of avascular osteonecrosis. Leukemia 2008, 22, 2054–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Cui, D.; Wang, B.; Tian, F.; Guo, L.; Yang, L.; Liu, B.; Yu, X. Treatment of early stage osteonecrosis of the femoral head with autologous implantation of bone marrow-derived and cultured mesenchymal stem cells. Bone 2012, 50, 325–330. [Google Scholar] [CrossRef]
- Daltro, G.C.; Fortuna, V.; de Souza, E.S.; Salles, M.M.; Carreira, A.C.; Meyer, R.; Freire, S.M.; Borojevic, R. Efficacy of autologous stem cell-based therapy for osteonecrosis of the femoral head in sickle cell disease: A five-year follow-up study. Stem Cell Res. Ther. 2015, 6110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangji, V.; De Maertelaer, V.; Hauzeur, J.P. Autologous bone marrow cell implantation in the treatment of non-traumatic osteonecrosis of the femoral head: Five year follow-up of a prospective controlled study. Bone 2011, 49, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Hauzeur, J.P.; De Maertelaer, V.; Baudoux, E.; Malaise, M.; Beguin, Y.; Gangji, V. Inefficacy of autologous bone marrow concentrate in stage three osteonecrosis: A randomized controlled double-blind trial. Int. Orthop. 2018, 42, 1429–1435. [Google Scholar] [CrossRef] [Green Version]
- Sen, R.K.; Tripathy, S.K.; Aggarwal, S.; Marwaha, N.; Sharma, R.R.; Khandelwal, N. Early results of core decompression and autologous bone marrow mononuclear cells instillation in femoral head osteonecrosis: A randomized control study. J. Arthroplast. 2012, 27, 679–686. [Google Scholar] [CrossRef]
- Hernigou, P.; Beaujean, F. Treatment of osteonecrosis with autologous bone marrow grafting. Clin. Orthop. Relat. Res. 2002, 405, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Hernigou, P.; Poignard, A.; Manicom, O.; Mathieu, G.; Rouard, H. The use of percutaneous autologous bone marrow transplantation in nonunion and avascular necrosis of bone. J. Bone Jt. Surg. Br. 2005, 87, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Tabatabaee, R.M.; Saberi, S.; Parvizi, J.; Mortazavi, S.M.; Farzan, M. Combining Concentrated Autologous Bone Marrow Stem Cells Injection With Core Decompression Improves Outcome for Patients with Early-Stage Osteonecrosis of the Femoral Head: A Comparative Study. J. Arthroplast. 2015, 30 (Suppl. 9), 11–15. [Google Scholar] [CrossRef]
- Martin, J.R.; Houdek, M.T.; Sierra, R.J. Use of concentrated bone marrow aspirate and platelet rich plasma during minimally invasive decompression of the femoral head in the treatment of osteonecrosis. Croat. Med. J. 2013, 54, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Civinini, R.; De Biase, P.; Carulli, C.; Matassi, F.; Nistri, L.; Capanna, R.; Innocenti, M. The use of an injectable calcium sulphate/calcium phosphate bioceramic in the treatment of osteonecrosis of the femoral head. Int. Orthop. 2012, 36, 1583–1588. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, Y.; Asada, R.; So, K.; Yonezawa, A.; Nankaku, M.; Mukai, K.; Ito-Ihara, T.; Tada, H.; Yamamoto, M.; Murayama, T.; et al. A pilot study of regenerative therapy using controlled release of recombinant human fibroblast growth factor for patients with pre-collapse osteonecrosis of the femoral head. Int. Orthop. 2016, 40, 1747–1754. [Google Scholar] [CrossRef]
- Rastogi, S.; Sankineani, S.R.; Nag, H.L.; Mohanty, S.; Shivanand, G.; Marimuthu, K.; Kumar, R.; Rijal, L. Intralesional autologous mesenchymal stem cells in management of osteonecrosis of femur: A preliminary study. Musculoskelet. Surg. 2013, 97, 223–228. [Google Scholar] [CrossRef]
- Pak, J. Autologous adipose tissue-derived stem cells induce persistent bone-like tissue in osteonecrotic femoral heads. Pain Physician 2012, 15, 75–85. [Google Scholar] [CrossRef]
- Cai, J.; Wu, Z.; Huang, L.; Chen, J.; Wu, C.; Wang, S.; Deng, Z.; Wu, W.; Luo, F.; Tan, J. Cotransplantation of bone marrow mononuclear cells and umbilical cord mesenchymal stem cells in avascular necrosis of the femoral head. Transpl. Proc. 2014, 46, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Wang, W.; Xu, T.; Zhang, S.; Xiao, L.; Chen, D.; Jin, H.; Tong, P. Combination treatment of biomechanical support and targeted intra-arterial infusion of peripheral blood stem cells mobilized by granulocyte-colony stimulating factor for the osteonecrosis of the femoral head: A randomized controlled clinical trial. J. Bone Min. Res. 2015, 30, 647–656. [Google Scholar] [CrossRef]
- Gomez-Barrena, E.; Padilla-Eguiluz, N.G.; Rosset, P.; Hernigou, P.; Baldini, N.; Ciapetti, G.; Gonzalo-Daganzo, R.M.; Avendano-Sola, C.; Rouard, H.; Giordano, R.; et al. Osteonecrosis of the Femoral Head Safely Healed with Autologous, Expanded, Bone Marrow-Derived Mesenchymal Stromal Cells in a Multicentric Trial with Minimum 5 Years Follow-Up. J. Clin. Med. 2021, 10, 508. [Google Scholar] [CrossRef]
- Hernigou, P.; Poignard, A.; Zilber, S.; Rouard, H. Cell therapy of hip osteonecrosis with autologous bone marrow grafting. Indian J. Orthop. 2009, 43, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, P.; El-Jawhari, J.J.; Giannoudis, P.V.; Burska, A.N.; Ponchel, F.; Jones, E.A. Age-related Changes in Bone Marrow Mesenchymal Stromal Cells: A Potential Impact on Osteoporosis and Osteoarthritis Development. Cell Transpl. 2017, 26, 1520–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.; Lee, Y.D.; Wagers, A.J. Stem cell aging: Mechanisms, regulators and therapeutic opportunities. Nat. Med. 2014, 20, 870–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.S.; Marban, E. Physiological levels of reactive oxygen species are required to maintain genomic stability in stem cells. Stem Cells 2010, 28, 1178–1185. [Google Scholar] [CrossRef] [Green Version]
- Jin, K. Modern Biological Theories of Aging. Aging Dis. 2010, 1, 72–74, PMC2995895. [Google Scholar]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Siegel, G.; Kluba, T.; Hermanutz-Klein, U.; Bieback, K.; Northoff, H.; Schafer, R. Phenotype, donor age and gender affect function of human bone marrow-derived mesenchymal stromal cells. BMC Med. 2013, 11146. [Google Scholar] [CrossRef] [Green Version]
- Kanda, Y.; Hinata, T.; Kang, S.W.; Watanabe, Y. Reactive oxygen species mediate adipocyte differentiation in mesenchymal stem cells. Life Sci. 2011, 89, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Baxter, M.A.; Wynn, R.F.; Jowitt, S.N.; Wraith, J.E.; Fairbairn, L.J.; Bellantuono, I. Study of telomere length reveals rapid aging of human marrow stromal cells following in vitro expansion. Stem Cells 2004, 22, 675–682. [Google Scholar] [CrossRef]
- Wagner, W.; Ho, A.D.; Zenke, M. Different facets of aging in human mesenchymal stem cells. Tissue Eng. Part. B Rev. 2010, 16, 445–453. [Google Scholar] [CrossRef]
- Yang, Y.K. Aging of mesenchymal stem cells: Implication in regenerative medicine. Regen. Ther. 2018, 9120–9122. [Google Scholar] [CrossRef]
- Cuthbert, R.; Boxall, S.A.; Tan, H.B.; Giannoudis, P.V.; McGonagle, D.; Jones, E. Single-platform quality control assay to quantify multipotential stromal cells in bone marrow aspirates prior to bulk manufacture or direct therapeutic use. Cytotherapy 2012, 14, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsov, S.A.; Mankani, M.H.; Gronthos, S.; Satomura, K.; Bianco, P.; Robey, P.G. Circulating skeletal stem cells. J. Cell Biol. 2001, 153, 1133–1140. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, S.A.; Mankani, M.H.; Leet, A.I.; Ziran, N.; Gronthos, S.; Robey, P.G. Circulating connective tissue precursors: Extreme rarity in humans and chondrogenic potential in guinea pigs. Stem Cells 2007, 25, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Fragkakis, E.M.; El-Jawhari, J.J.; Dunsmuir, R.A.; Millner, P.A.; Rao, A.S.; Henshaw, K.T.; Pountos, I.; Jones, E.; Giannoudis, P.V. Vertebral body versus iliac crest bone marrow as a source of multipotential stromal cells: Comparison of processing techniques, tri-lineage differentiation and application on a scaffold for spine fusion. PLoS ONE 2018, 13, e0197969. [Google Scholar] [CrossRef]
- Dal Pozzo, S.; Urbani, S.; Mazzanti, B.; Luciani, P.; Deledda, C.; Lombardini, L.; Benvenuti, S.; Peri, A.; Bosi, A.; Saccardi, R. High recovery of mesenchymal progenitor cells with non-density gradient separation of human bone marrow. Cytotherapy 2010, 12, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Horn, P.; Bork, S.; Wagner, W. Standardized isolation of human mesenchymal stromal cells with red blood cell lysis. Methods Mol. Biol. 2011, 69823–69835. [Google Scholar] [CrossRef]
- Stenderup, K.; Justesen, J.; Eriksen, E.F.; Rattan, S.I.; Kassem, M. Number and proliferative capacity of osteogenic stem cells are maintained during aging and in patients with osteoporosis. J. Bone Min. Res. 2001, 16, 1120–1129. [Google Scholar] [CrossRef]
- Muschler, G.F.; Nitto, H.; Boehm, C.A.; Easley, K.A. Age- and gender-related changes in the cellularity of human bone marrow and the prevalence of osteoblastic progenitors. J. Orthop. Res. 2001, 19, 117–125. [Google Scholar] [CrossRef]
- Stolzing, A.; Jones, E.; McGonagle, D.; Scutt, A. Age-related changes in human bone marrow-derived mesenchymal stem cells: Consequences for cell therapies. Mech. Ageing Dev. 2008, 129, 163–173. [Google Scholar] [CrossRef]
- Kuznetsov, S.A.; Mankani, M.H.; Bianco, P.; Robey, P.G. Enumeration of the colony-forming units-fibroblast from mouse and human bone marrow in normal and pathological conditions. Stem Cell Res. 2009, 2, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wong, W.H.; Chan, S.; Chim, J.C.; Cheung, K.M.; Lee, T.L.; Au, W.Y.; Ha, S.Y.; Lie, A.K.; Lau, Y.L.; et al. Factors affecting mesenchymal stromal cells yield from bone marrow aspiration. Chin. J. Cancer Res. 2011, 23, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Jawhari, J.J.; Cuthbert, R.; McGonagle, D.; Jones, E.; Giannoudis, P.V. The CD45lowCD271high Cell Prevalence in Bone Marrow Samples May Provide a Useful Measurement of the Bone Marrow Quality for Cartilage and Bone Regenerative Therapy. J. Bone Jt. Surg. Am. 2017, 99, 1305–1313. [Google Scholar] [CrossRef]
- Ganguly, P.; El-Jawhari, J.J.; Burska, A.N.; Ponchel, F.; Giannoudis, P.V.; Jones, E.A. The Analysis of In Vivo Aging in Human Bone Marrow Mesenchymal Stromal Cells Using Colony-Forming Unit-Fibroblast Assay and the CD45(low)CD271(+) Phenotype. Stem Cells Int. 2019, 20195197983. [Google Scholar] [CrossRef] [Green Version]
- Rebolj, K.; Veber, M.; Drobnic, M.; Malicev, E. Hematopoietic stem cell and mesenchymal stem cell population size in bone marrow samples depends on patient’s age and harvesting technique. Cytotechnology 2018, 70, 1575–1583. [Google Scholar] [CrossRef]
- Harichandan, A.; Buhring, H.J. Prospective isolation of human MSC. Best Pract. Res. Clin. Haematol. 2011, 24, 25–36. [Google Scholar] [CrossRef]
- Li, H.; Ghazanfari, R.; Zacharaki, D.; Lim, H.C.; Scheding, S. Isolation and characterization of primary bone marrow mesenchymal stromal cells. Ann. N. Y. Acad. Sci. 2016, 1370, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Kouroupis, D.; Sanjurjo-Rodriguez, C.; Jones, E.; Correa, D. Mesenchymal Stem Cell Functionalization for Enhanced Therapeutic Applications. Tissue Eng. Part. B Rev. 2019, 25, 55–77. [Google Scholar] [CrossRef]
- Josephson, A.M.; Bradaschia-Correa, V.; Lee, S.; Leclerc, K.; Patel, K.S.; Muinos Lopez, E.; Litwa, H.P.; Neibart, S.S.; Kadiyala, M.; Wong, M.Z.; et al. Age-related inflammation triggers skeletal stem/progenitor cell dysfunction. Proc. Natl. Acad. Sci. USA 2019, 116, 6995–7004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zha, K.; Yang, Y.; Tian, G.; Sun, Z.; Yang, Z.; Li, X.; Sui, X.; Liu, S.; Zhao, J.; Guo, Q. Nerve growth factor (NGF) and NGF receptors in mesenchymal stem/stromal cells: Impact on potential therapies. Stem Cells Transl. Med. 2021. [Google Scholar] [CrossRef]
- Ganguly, P.; Burska, A.N.; Davis, C.L.M.; El-Jawhari, J.J.; Giannoudis, P.V.; Jones, E.A. Intrinsic Type 1 Interferon (IFN1) Profile of Uncultured Human Bone Marrow CD45(low)CD271(+) Multipotential Stromal Cells (BM-MSCs): The Impact of Donor Age, Culture Expansion and IFNalpha and IFNbeta Stimulation. Biomedicines 2020, 8, 214. [Google Scholar] [CrossRef] [PubMed]
- Block, T.J.; Marinkovic, M.; Tran, O.N.; Gonzalez, A.O.; Marshall, A.; Dean, D.D.; Chen, X.D. Restoring the quantity and quality of elderly human mesenchymal stem cells for autologous cell-based therapies. Stem Cell Res. Ther. 2017, 8, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrzejewska, A.; Catar, R.; Schoon, J.; Qazi, T.H.; Sass, F.A.; Jacobi, D.; Blankenstein, A.; Reinke, S.; Krüger, D.; Streitz, M.; et al. Multi-Parameter Analysis of Biobanked Human Bone Marrow Stromal Cells Shows Little Influence for Donor Age and Mild Comorbidities on Phenotypic and Functional Properties. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- El-Jawhari, J.J.; Kleftouris, G.; El-Sherbiny, Y.; Saleeb, H.; West, R.M.; Jones, E.; Giannoudis, P.V. Defective Proliferation and Osteogenic Potential with Altered Immunoregulatory Phenotype of Native Bone Marrow-Multipotential Stromal Cells in Atrophic Fracture Non-Union. Sci. Rep. 2019, 9, 17340. [Google Scholar] [CrossRef]
- Rahmati, M.; Nalesso, G.; Mobasheri, A.; Mozafari, M. Aging and osteoarthritis: Central role of the extracellular matrix. Ageing Res. Rev. 2017, 4020–4030. [Google Scholar] [CrossRef]
- Buckwalter, J.A.; Woo, S.L.; Goldberg, V.M.; Hadley, E.C.; Booth, F.; Oegema, T.R.; Eyre, D.R. Soft-tissue aging and musculoskeletal function. J. Bone Jt. Surg. Am. 1993, 75, 1533–1548. [Google Scholar] [CrossRef]
- Mitani, H.; Takahashi, I.; Onodera, K.; Bae, J.W.; Sato, T.; Takahashi, N.; Sasano, Y.; Igarashi, K.; Mitani, H. Comparison of age-dependent expression of aggrecan and ADAMTSs in mandibular condylar cartilage, tibial growth plate, and articular cartilage in rats. Histochem. Cell Biol. 2006, 126, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Germaschewski, F.M.; Matheny, C.J.; Larkin, J.; Liu, F.; Thomas, L.R.; Saunders, J.S.; Sully, K.; Whittall, C.; Boyle, Y.; Peters, G.; et al. Quantitation OF ARGS aggrecan fragments in synovial fluid, serum and urine from osteoarthritis patients. Osteoarthr. Cartil. 2014, 22, 690–697. [Google Scholar] [CrossRef] [Green Version]
- Forsyth, C.B.; Cole, A.; Murphy, G.; Bienias, J.L.; Im, H.J.; Loeser, R.F.J. Increased matrix metalloproteinase-13 production with aging by human articular chondrocytes in response to catabolic stimuli. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 1118–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, L.H.; Lai, W.F.; Chang, S.F.; Wong, C.C.; Fan, C.Y.; Fang, C.L.; Tsai, Y.H. The effect of type II collagen on MSC osteogenic differentiation and bone defect repair. Biomaterials 2014, 35, 2680–2691. [Google Scholar] [CrossRef]
- Yu, C.; Peall, I.W.; Pham, S.H.; Okolicsanyi, R.K.; Griffiths, L.R.; Haupt, L.M. Syndecan-1 Facilitates the Human Mesenchymal Stem Cell Osteo-Adipogenic Balance. Int. J. Mol. Sci. 2020, 21, 3884. [Google Scholar] [CrossRef]
- Atesok, K.; Fu, F.H.; Sekiya, I.; Stolzing, A.; Ochi, M.; Rodeo, S.A. Stem cells in degenerative orthopaedic pathologies: Effects of aging on therapeutic potential. Knee Surg. Sports Traumatol. Arthrosc. 2017, 25, 626–636. [Google Scholar] [CrossRef]
- Lynch, K.; Pei, M. Age associated communication between cells and matrix: A potential impact on stem cell-based tissue regeneration strategies. Organogenesis 2014, 10, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Li, W.; Lu, Z.; Chen, R.; Ling, J.; Ran, Q.; Jilka, R.L.; Chen, X.D. Rescuing replication and osteogenesis of aged mesenchymal stem cells by exposure to a young extracellular matrix. FASEB J. 2011, 25, 1474–1485. [Google Scholar] [CrossRef] [Green Version]
- Canalis, E. Mechanisms of glucocorticoid-induced osteoporosis. Curr. Opin. Rheumatol. 2003, 15, 454–457. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.H.; Wang, X.L.; Yang, H.L.; Zhao, D.W.; Qin, L. Steroid-associated osteonecrosis: Epidemiology, pathophysiology, animal model, prevention, and potential treatments (an overview). J. Orthop. Transl. 2015, 3, 58–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, R.S. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol. Metab. Clin. N. Am. 2012, 41, 595–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerachian, M.A.; Seguin, C.; Harvey, E.J. Glucocorticoids in osteonecrosis of the femoral head: A new understanding of the mechanisms of action. J. Steroid Biochem. Mol. Biol. 2009, 114, 121–128. [Google Scholar] [CrossRef]
- Chang, J.K.; Ho, M.L.; Yeh, C.H.; Chen, C.H.; Wang, G.J. Osteogenic gene expression decreases in stromal cells of patients with osteonecrosis. Clin. Orthop. Relat. Res. 2006, 453, 286–292. [Google Scholar] [CrossRef]
- Hong, L.; Sultana, H.; Paulius, K.; Zhang, G. Steroid regulation of proliferation and osteogenic differentiation of bone marrow stromal cells: A gender difference. J. Steroid Biochem. Mol. Biol. 2009, 114, 180–185. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Cen, L.; Zhou, H.; Yin, S.; Liu, G.; Liu, W.; Cao, Y.; Cui, L. The role of the extracellular signal-related kinase signaling pathway in osteogenic differentiation of human adipose-derived stem cells and in adipogenic transition initiated by dexamethasone. Tissue Eng. Part. A 2009, 15, 3487–3497. [Google Scholar] [CrossRef] [Green Version]
- Fadini, G.P.; de Kreutzenberg, S.; Albiero, M.; Coracina, A.; Pagnin, E.; Baesso, I.; Cignarella, A.; Bolego, C.; Plebani, M.; Nardelli, G.B.; et al. Gender differences in endothelial progenitor cells and cardiovascular risk profile: The role of female estrogens. Arter. Thromb. Vasc. Biol. 2008, 28, 997–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, B.B.; Moses, L.F. Sex differences in the distribution and regulation of glucocorticoid receptors in cardiac tissues of rats. J. Mol. Cell Cardiol. 1986, 18, 331–333. [Google Scholar] [CrossRef]
- Nuzzi, R.; Gunetti, M.; Rustichelli, D.; Roagna, B.; Fronticelli Bardelli, F.; Fagioli, F.; Ferrero, I. Effect of In Vitro Exposure of Corticosteroid Drugs, Conventionally Used in AMD Treatment, on Mesenchymal Stem Cells. Stem Cells Int. 2012, 2012946090. [Google Scholar] [CrossRef]
- Kato, T.; Khanh, V.C.; Sato, K.; Kimura, K.; Yamashita, T.; Sugaya, H.; Yoshioka, T.; Mishima, H.; Ohneda, O. Elevated Expression of Dkk-1 by Glucocorticoid Treatment Impairs Bone Regenerative Capacity of Adipose Tissue-Derived Mesenchymal Stem Cells. Stem Cells Dev. 2018, 27, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Yang, S.; Xu, W.; Shen, J.K.; Ye, S.; Liu, X.; Dong, Z.; Xiao, B.; Feng, Y. MiR-708 promotes steroid-induced osteonecrosis of femoral head, suppresses osteogenic differentiation by targeting SMAD3. Sci. Rep. 2016, 622599. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gan, Y.; Li, W.; Su, J.; Zhang, Y.; Huang, Y.; Roberts, A.I.; Han, Y.; Li, J.; Wang, Y.; et al. The interaction between mesenchymal stem cells and steroids during inflammation. Cell Death Dis. 2014, 5, e1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovach, T.K.; Dighe, A.S.; Lobo, P.I.; Cui, Q. Interactions between MSCs and immune cells: Implications for bone healing. J. Immunol. Res. 2015, 2015752510. [Google Scholar] [CrossRef]
- Lienau, J.; Schmidt-Bleek, K.; Peters, A.; Weber, H.; Bail, H.J.; Duda, G.N.; Perka, C.; Schell, H. Insight into the molecular pathophysiology of delayed bone healing in a sheep model. Tissue Eng. Part. A 2010, 16, 191–199. [Google Scholar] [CrossRef]
- Hoff, P.; Gaber, T.; Schmidt-Bleek, K.; Senturk, U.; Tran, C.L.; Blankenstein, K.; Lutkecosmann, S.; Bredahl, J.; Schuler, H.J.; Simon, P.; et al. Immunologically restricted patients exhibit a pronounced inflammation and inadequate response to hypoxia in fracture hematomas. Immunol. Res. 2011, 51, 116–122. [Google Scholar] [CrossRef]
- Li, T.; Liu, Z.L.; Xiao, M.; Yang, Z.Z.; Peng, M.Z.; Li, C.D.; Zhou, X.J.; Wang, J.W. Impact of bone marrow mesenchymal stem cell immunomodulation on the osteogenic effects of laponite. Stem Cell Res. Ther. 2018, 9, 100. [Google Scholar] [CrossRef] [Green Version]
- Granero-Molto, F.; Weis, J.A.; Miga, M.I.; Landis, B.; Myers, T.J.; O’Rear, L.; Longobardi, L.; Jansen, E.D.; Mortlock, D.P.; Spagnoli, A. Regenerative effects of transplanted mesenchymal stem cells in fracture healing. Stem Cells 2009, 27, 1887–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, B.D.; Chen, J.; Zhang, X.Y.; He, T.; Zhao, P.; Zheng, C.X.; Li, M.; Hu, C.H.; Jin, Y. Gender-independent efficacy of mesenchymal stem cell therapy in sex hormone-deficient bone loss via immunosuppression and resident stem cell recovery. Exp. Mol. Med. 2018, 50, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernigou, P.; Beaujean, F.; Lambotte, J.C. Decrease in the mesenchymal stem-cell pool in the proximal femur in corticosteroid-induced osteonecrosis. J. Bone Jt. Surg. Br. 1999, 81, 349–355. [Google Scholar] [CrossRef]
- Brabnikova Maresova, K.; Pavelka, K.; Stepan, J.J. Acute effects of glucocorticoids on serum markers of osteoclasts, osteoblasts, and osteocytes. Calcif. Tissue Int. 2013, 92, 354–361. [Google Scholar] [CrossRef]
- Stewart, P.M. Tissue-specific Cushing’s syndrome, 11beta-hydroxysteroid dehydrogenases and the redefinition of corticosteroid hormone action. Eur. J. Endocrinol. 2003, 149, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.Z.; Li, H.; Tao, Y.Q.; Zhou, X.P.; Yang, Z.R.; Xiao, Y.X.; Li, F.C.; Han, B.; Chen, Q.X. Dual delivery for stem cell differentiation using dexamethasone and bFGF in/on polymeric microspheres as a cell carrier for nucleus pulposus regeneration. J. Mater. Sci. Mater. Med. 2012, 23, 1097–1107. [Google Scholar] [CrossRef]
- Walsh, J.S.; Eastell, R. Osteoporosis in men. Nat. Rev. Endocrinol. 2013, 9, 637–645. [Google Scholar] [CrossRef]
- Maurel, D.B.; Boisseau, N.; Benhamou, C.L.; Jaffre, C. Alcohol and bone: Review of dose effects and mechanisms. Osteoporos. Int. 2012, 23, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Seamon, J.; Keller, T.; Saleh, J.; Cui, Q. The pathogenesis of nontraumatic osteonecrosis. Arthritis 2012, 2012601763. [Google Scholar] [CrossRef] [Green Version]
- Aldahmash, A. Skeletal stem cells and their contribution to skeletal fragility: Senescence and rejuvenation. Biogerontology 2016, 17, 297–304. [Google Scholar] [CrossRef]
- Bonyadi, M.; Waldman, S.D.; Liu, D.; Aubin, J.E.; Grynpas, M.D.; Stanford, W.L. Mesenchymal progenitor self-renewal deficiency leads to age-dependent osteoporosis in Sca-1/Ly-6A null mice. Proc. Natl. Acad. Sci. USA 2003, 100, 5840–5845. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, M.; Yan, J.; Liu, T.; Pan, G.; Yang, H.; Pei, M.; He, F. Alcohol Induces Cellular Senescence and Impairs Osteogenic Potential in Bone Marrow-Derived Mesenchymal Stem Cells. Alcohol. Alcohol. 2017, 52, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Huff, N.K.; Spencer, N.D.; Gimble, J.M.; Bagby, G.J.; Nelson, S.; Lopez, M.J. Impaired expansion and multipotentiality of adult stromal cells in a rat chronic alcohol abuse model. Alcohol 2011, 45, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Vassallo, P.F.; Simoncini, S.; Ligi, I.; Chateau, A.L.; Bachelier, R.; Robert, S.; Morere, J.; Fernandez, S.; Guillet, B.; Marcelli, M.; et al. Accelerated senescence of cord blood endothelial progenitor cells in premature neonates is driven by SIRT1 decreased expression. Blood 2014, 123, 2116–2126. [Google Scholar] [CrossRef]
- Liu, Y.; Kou, X.; Chen, C.; Yu, W.; Su, Y.; Kim, Y.; Shi, S.; Liu, Y. Chronic High Dose Alcohol Induces Osteopenia via Activation of mTOR Signaling in Bone Marrow Mesenchymal Stem Cells. Stem Cells 2016, 34, 2157–2168. [Google Scholar] [CrossRef]
- Gong, Z.; Wezeman, F.H. Inhibitory effect of alcohol on osteogenic differentiation in human bone marrow-derived mesenchymal stem cells. Alcohol Clin. Exp. Res. 2004, 28, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Lauing, K.L.; Roper, P.M.; Nauer, R.K.; Callaci, J.J. Acute alcohol exposure impairs fracture healing and deregulates beta-catenin signaling in the fracture callus. Alcohol Clin. Exp. Res. 2012, 36, 2095–2103. [Google Scholar] [CrossRef] [Green Version]
- Elmali, N.; Ertem, K.; Ozen, S.; Inan, M.; Baysal, T.; Guner, G.; Bora, A. Fracture healing and bone mass in rats fed on liquid diet containing ethanol. Alcohol Clin. Exp. Res. 2002, 26, 509–513. [Google Scholar] [CrossRef]
- Nyquist, F.; Halvorsen, V.; Madsen, J.E.; Nordsletten, L.; Obrant, K.J. Ethanol and its effects on fracture healing and bone mass in male rats. Acta Orthop. Scand. 1999, 70, 212–216. [Google Scholar] [CrossRef] [Green Version]
- Perrien, D.S.; Wahl, E.C.; Hogue, W.R.; Feige, U.; Aronson, J.; Ronis, M.J.; Badger, T.M.; Lumpkin, C.K.J. IL-1 and TNF antagonists prevent inhibition of fracture healing by ethanol in rats. Toxicol. Sci. 2004, 82, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, P.C.; Hou, S.M.; Chen, R.J.; Peng, H.W.; Hsieh, C.F.; Kuo, M.L.; Yen, M.L. Resveratrol promotes osteogenesis of human mesenchymal stem cells by upregulating RUNX2 gene expression via the SIRT1/FOXO3A axis. J. Bone Min. Res. 2011, 26, 2552–2563. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.K.; Callaci, J.J.; Lauing, K.L.; Otis, J.S.; Radek, K.A.; Jones, M.K.; Kovacs, E.J. Alcohol exposure and mechanisms of tissue injury and repair. Alcohol Clin. Exp. Res. 2011, 35, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Callaci, J.J.; Juknelis, D.; Patwardhan, A.; Wezeman, F.H. Binge alcohol treatment increases vertebral bone loss following ovariectomy: Compensation by intermittent parathyroid hormone. Alcohol Clin. Exp. Res. 2006, 30, 665–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaci, J.J.; Juknelis, D.; Patwardhan, A.; Sartori, M.; Frost, N.; Wezeman, F.H. The effects of binge alcohol exposure on bone resorption and biomechanical and structural properties are offset by concurrent bisphosphonate treatment. Alcohol Clin. Exp. Res. 2004, 28, 182–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaci, J.J.; Himes, R.; Lauing, K.; Wezeman, F.H.; Brownson, K. Binge alcohol-induced bone damage is accompanied by differential expression of bone remodeling-related genes in rat vertebral bone. Calcif. Tissue Int. 2009, 84, 474–484. [Google Scholar] [CrossRef] [Green Version]
- Callaci, J.J.; Himes, R.; Lauing, K.; Roper, P. Long-term modulations in the vertebral transcriptome of adolescent-stage rats exposed to binge alcohol. Alcohol Alcohol. 2010, 45, 332–346. [Google Scholar] [CrossRef] [PubMed]
- Lauing, K.L.; Sundaramurthy, S.; Nauer, R.K.; Callaci, J.J. Exogenous activation of Wnt/beta-catenin signaling attenuates binge alcohol-induced deficient bone fracture healing. Alcohol Alcohol. 2014, 49, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Himes, R.; Wezeman, F.H.; Callaci, J.J. Identification of novel bone-specific molecular targets of binge alcohol and ibandronate by transcriptome analysis. Alcohol Clin. Exp. Res. 2008, 32, 1167–1180. [Google Scholar] [CrossRef] [Green Version]
- Driver, J.; Weber, C.E.; Callaci, J.J.; Kothari, A.N.; Zapf, M.A.; Roper, P.M.; Borys, D.; Franzen, C.A.; Gupta, G.N.; Wai, P.Y.; et al. Alcohol inhibits osteopontin-dependent transforming growth factor-beta1 expression in human mesenchymal stem cells. J. Biol. Chem. 2015, 290, 9959–9973. [Google Scholar] [CrossRef] [Green Version]
- Natoli, R.M.; Yu, H.; Meislin, M.C.; Abbasnia, P.; Roper, P.; Vuchkovska, A.; Xiao, X.; Stock, S.R.; Callaci, J.J. Alcohol exposure decreases osteopontin expression during fracture healing and osteopontin-mediated mesenchymal stem cell migration in vitro. J. Orthop. Surg. Res. 2018, 13, 101. [Google Scholar] [CrossRef] [Green Version]
- Sears, B.W.; Volkmer, D.; Yong, S.; Himes, R.D.; Lauing, K.; Morgan, M.; Stover, M.D.; Callaci, J.J. Binge alcohol exposure modulates rodent expression of biomarkers of the immunoinflammatory response to orthopaedic trauma. J. Bone Jt. Surg. Am. 2011, 93, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Zou, C.; Luo, Q.; Qin, J.; Shi, Y.; Yang, L.; Ju, B.; Song, G. Osteopontin promotes mesenchymal stem cell migration and lessens cell stiffness via integrin beta1, FAK, and ERK pathways. Cell Biochem. Biophys. 2013, 65, 455–462. [Google Scholar] [CrossRef]
- Kumagai, H.; Yoshioka, T.; Sugaya, H.; Tomaru, Y.; Shimizu, Y.; Yamazaki, M.; Mishima, H. Quantitative assessment of mesenchymal stem cells contained in concentrated autologous bone marrow aspirate transplantation for the treatment of osteonecrosis of the femoral head: Predictive factors and differences by etiology. BMC Res. Notes 2018, 11, 848. [Google Scholar] [CrossRef]
- Volkmer, D.L.; Sears, B.; Lauing, K.L.; Nauer, R.K.; Roper, P.M.; Yong, S.; Stover, M.; Callaci, J.J. Antioxidant therapy attenuates deficient bone fracture repair associated with binge alcohol exposure. J. Orthop. Trauma 2011, 25, 516–521. [Google Scholar] [CrossRef] [Green Version]
- Wezeman, F.H.; Juknelis, D.; Himes, R.; Callaci, J.J. Vitamin D and ibandronate prevent cancellous bone loss associated with binge alcohol treatment in male rats. Bone 2007, 41, 639–645. [Google Scholar] [CrossRef] [Green Version]
- Peris, P.; Pares, A.; Guanabens, N.; Del Rio, L.; Pons, F.; Martinez de Osaba, M.J.; Monegal, A.; Caballeria, J.; Rodes, J.; Munoz-Gomez, J. Bone mass improves in alcoholics after 2 years of abstinence. J. Bone Min. Res. 1994, 9, 1607–1612. [Google Scholar] [CrossRef]
- Almeida, A.; Roberts, I. Bone involvement in sickle cell disease. Br. J. Haematol. 2005, 129, 482–490. [Google Scholar] [CrossRef]
- Adesina, O.; Brunson, A.; Keegan, T.H.M.; Wun, T. Osteonecrosis of the femoral head in sickle cell disease: Prevalence, comorbidities, and surgical outcomes in California. Blood Adv. 2017, 1, 1287–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernigou, P.; Flouzat-Lachaniette, C.H.; Daltro, G.; Galacteros, F. Talar Osteonecrosis Related to Adult Sickle Cell Disease: Natural Evolution from Early to Late Stages. J. Bone Jt. Surg. Am. 2016, 98, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Poignard, A.; Flouzat-Lachaniette, C.H.; Amzallag, J.; Galacteros, F.; Hernigou, P. The natural progression of symptomatic humeral head osteonecrosis in adults with sickle cell disease. J. Bone Jt. Surg. Am. 2012, 94, 156–162. [Google Scholar] [CrossRef] [Green Version]
- Stenger, E.O.; Chinnadurai, R.; Yuan, S.; Garcia, M.; Arafat, D.; Gibson, G.; Krishnamurti, L.; Galipeau, J. Bone Marrow-Derived Mesenchymal Stromal Cells from Patients with Sickle Cell Disease Display Intact Functionality. Biol. Blood Marrow Transpl. 2017, 23, 736–745. [Google Scholar] [CrossRef]
- Ribeiro, T.O.; Silveira, B.M.; Meira, M.C.; Carreira, A.C.O.; Sogayar, M.C.; Meyer, R.; Fortuna, V. Investigating the potential of the secretome of mesenchymal stem cells derived from sickle cell disease patients. PLoS ONE 2019, 14, e0222093. [Google Scholar] [CrossRef]
- Ribeiro, T.O.; Daltro, P.B.; Daltro, G.C.; Freire, S.M.; Meyer, R.; Fortuna, V. Quantification and Comprehensive Analysis of Mesenchymal Stromal Cells in Bone Marrow Samples from Sickle Cell Disease Patients with Osteonecrosis. Stem Cells Int. 2020, 20208841191. [Google Scholar] [CrossRef]
- Gildásio Daltro, N.d.S.S.R.; Saide Maria Sarmento Trindade Paulo Borges, I.C.d.A.a.S.M. Pediatric Sickle Cell Disease Osteonecrosis of the Femoral Head: A Treatment Proposal. Clin. Pediatrics 2020, 3, 1022. [Google Scholar]
- Hernigou, P.; Bernaudin, F.; Reinert, P.; Kuentz, M.; Vernant, J.P. Bone-marrow transplantation in sickle-cell disease. Effect on osteonecrosis: A case report with a four-year follow-up. J. Bone Jt. Surg. Am. 1997, 79, 1726–1730. [Google Scholar] [CrossRef] [PubMed]
- Linari, S.; Castaman, G. Clinical manifestations and management of Gaucher disease. Clin. Cases Min. Bone Metab. 2015, 12, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Katz, K.; Horev, G.; Grunebaum, M.; Yosipovitch, Z. The natural history of osteonecrosis of the femoral head in children and adolescents who have Gaucher disease. J. Bone Jt. Surg. Am. 1996, 78, 14–19. [Google Scholar] [CrossRef]
- Mistry, P.K.; Liu, J.; Yang, M.; Nottoli, T.; McGrath, J.; Jain, D.; Zhang, K.; Keutzer, J.; Chuang, W.L.; Mehal, W.Z.; et al. Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc. Natl. Acad. Sci. USA 2010, 107, 19473–19478. [Google Scholar] [CrossRef] [Green Version]
- Campeau, P.M.; Rafei, M.; Boivin, M.N.; Sun, Y.; Grabowski, G.A.; Galipeau, J. Characterization of Gaucher disease bone marrow mesenchymal stromal cells reveals an altered inflammatory secretome. Blood 2009, 114, 3181–3190. [Google Scholar] [CrossRef] [Green Version]
- Lecourt, S.; Mouly, E.; Freida, D.; Cras, A.; Ceccaldi, R.; Heraoui, D.; Chomienne, C.; Marolleau, J.P.; Arnulf, B.; Porcher, R.; et al. A prospective study of bone marrow hematopoietic and mesenchymal stem cells in type 1 Gaucher disease patients. PLoS ONE 2013, 8, e69293. [Google Scholar] [CrossRef]
- Gladman, D.D.; Urowitz, M.B.; Chaudhry-Ahluwalia, V.; Hallet, D.C.; Cook, R.J. Predictive factors for symptomatic osteonecrosis in patients with systemic lupus erythematosus. J. Rheumatol. 2001, 28, 761–765. [Google Scholar] [PubMed]
- Calvo-Alen, J.; McGwin, G.; Toloza, S.; Fernandez, M.; Roseman, J.M.; Bastian, H.M.; Cepeda, E.J.; Gonzalez, E.B.; Baethge, B.A.; Fessler, B.J.; et al. Systemic lupus erythematosus in a multiethnic US cohort (LUMINA): XXIV. Cytotoxic treatment is an additional risk factor for the development of symptomatic osteonecrosis in lupus patients: Results of a nested matched case-control study. Ann. Rheum. Dis. 2006, 65, 785–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorraji, S.E.; Hovd, A.K.; Kanapathippillai, P.; Bakland, G.; Eilertsen, G.O.; Figenschau, S.L.; Fenton, K.A. Mesenchymal stem cells and T cells in the formation of Tertiary Lymphoid Structures in Lupus Nephritis. Sci. Rep. 2018, 8, 7861. [Google Scholar] [CrossRef] [PubMed]
- Che, N.; Li, X.; Zhang, L.; Liu, R.; Chen, H.; Gao, X.; Shi, S.; Chen, W.; Sun, L. Impaired B cell inhibition by lupus bone marrow mesenchymal stem cells is caused by reduced CCL2 expression. J. Immunol. 2014, 193, 5306–5314. [Google Scholar] [CrossRef]
- Ji, J.; Wu, Y.; Meng, Y.; Zhang, L.; Feng, G.; Xia, Y.; Xue, W.; Zhao, S.; Gu, Z.; Shao, X. JAK-STAT signaling mediates the senescence of bone marrow-mesenchymal stem cells from systemic lupus erythematosus patients. Acta Biochim. Biophys. Sin. 2017, 49, 208–215. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Tan, W.; Feng, G.; Meng, Y.; Shen, B.; Liu, H.; Cheng, C. Wnt/beta-catenin signaling mediates the senescence of bone marrow-mesenchymal stem cells from systemic lupus erythematosus patients through the p53/p21 pathway. Mol. Cell Biochem. 2014, 387, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Gu, Z.; Shen, B.; Jiang, J.; Meng, Y.; Da, Z.; Liu, H.; Tao, T.; Cheng, C. PTEN/Akt-p27(kip1) Signaling Promote the BM-MSCs Senescence and Apoptosis in SLE Patients. J. Cell Biochem. 2015, 116, 1583–1594. [Google Scholar] [CrossRef]
- Nie, Y.; Lau, C.S.; Lie, A.K.; Chan, G.C.; Mok, M.Y. Defective phenotype of mesenchymal stem cells in patients with systemic lupus erythematosus. Lupus 2010. [Google Scholar] [CrossRef]
- Sun, L.Y.; Zhang, H.Y.; Feng, X.B.; Hou, Y.Y.; Lu, L.W.; Fan, L.M. Abnormality of bone marrow-derived mesenchymal stem cells in patients with systemic lupus erythematosus. Lupus 2007, 16, 121–128. [Google Scholar] [CrossRef]
- Lourenco, E.V.; La Cava, A. Cytokines in systemic lupus erythematosus. Curr. Mol. Med. 2009, 9, 242–254. [Google Scholar] [CrossRef] [Green Version]
- Fathollahi, A.; Gabalou, N.B.; Aslani, S. Mesenchymal stem cell transplantation in systemic lupus erythematous, a mesenchymal stem cell disorder. Lupus 2018, 27, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Li, X.; Chen, H.; Che, N.; Zhou, S.; Lu, Z.; Shi, S.; Sun, L. High level of reactive oxygen species impaired mesenchymal stem cell migration via overpolymerization of F-actin cytoskeleton in systemic lupus erythematosus. Pathol. Biol. 2014, 62, 382–390. [Google Scholar] [CrossRef]
- Geng, L.; Tang, X.; Zhou, K.; Wang, D.; Wang, S.; Yao, G.; Chen, W.; Gao, X.; Chen, W.; Shi, S.; et al. MicroRNA-663 induces immune dysregulation by inhibiting TGF-beta1 production in bone marrow-derived mesenchymal stem cells in patients with systemic lupus erythematosus. Cell Mol. Immunol. 2019, 16, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Mardones, R.; Camacho, D.; Monsalvo, F.; Zulch, N.; Jofre, C.; Minguell, J.J. Treatment of osteonecrosis of the femoral head by core decompression and implantation of fully functional ex vivo-expanded bone marrow-derived mesenchymal stem cells: A proof-of-concept study. Stem Cells Cloning 2019, 1211–1216. [Google Scholar] [CrossRef] [Green Version]
- Peer, A.; Khamaisi, M. Diabetes as a risk factor for medication-related osteonecrosis of the jaw. J. Dent. Res. 2015, 94, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Tevlin, R.; Seo, E.Y.; Marecic, O.; McArdle, A.; Tong, X.; Zimdahl, B.; Malkovskiy, A.; Sinha, R.; Gulati, G.; Li, X.; et al. Pharmacological rescue of diabetic skeletal stem cell niches. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Han, J.W.; Lee, J.Y.; Choi, Y.J.; Sohn, Y.D.; Song, M.; Yoon, Y.S. Diabetic Mesenchymal Stem Cells Are Ineffective for Improving Limb Ischemia Due to Their Impaired Angiogenic Capability. Cell Transpl. 2015, 24, 1571–1584. [Google Scholar] [CrossRef] [Green Version]
- Roszer, T.; Jozsa, T.; Kiss-Toth, E.D.; De Clerck, N.; Balogh, L. Leptin receptor deficient diabetic (db/db) mice are compromised in postnatal bone regeneration. Cell Tissue Res. 2014, 356, 195–206. [Google Scholar] [CrossRef]
- Fontaine, J.L.; Hunt, N.A.; Curry, S.; Kearney, T.; Jupiter, D.; Shibuya, N.; Lavery, L.A. Fracture healing and biomarker expression in a diabetic Zucker rat model. J. Am. Podiatr. Med. Assoc. 2014, 104, 428–433. [Google Scholar] [CrossRef]
- Chen, Q.Q.; Wang, W.M. Expression of FGF-2 and IGF-1 in diabetic rats with fracture. Asian Pac. J. Trop. Med. 2014, 7, 71–75. [Google Scholar] [CrossRef]
- Hamann, C.; Goettsch, C.; Mettelsiefen, J.; Henkenjohann, V.; Rauner, M.; Hempel, U.; Bernhardt, R.; Fratzl-Zelman, N.; Roschger, P.; Rammelt, S.; et al. Delayed bone regeneration and low bone mass in a rat model of insulin-resistant type 2 diabetes mellitus is due to impaired osteoblast function. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E1220-8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Chen, X.; Liu, B.; Chou, K.; Liu, Z.; Deng, J. Expression of PPARgamma and Cbfalpha1 mRNA in bone marrow cells in Type 2 diabetic rats and its correlation with impaired fracture healing. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2009, 34, 957–964. [Google Scholar]
- Roy, B. Biomolecular basis of the role of diabetes mellitus in osteoporosis and bone fractures. World J. Diabetes 2013, 4, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Hamada, Y.; Fukagawa, M. Bone formation in spontaneously diabetic Torii-newly established model of non-obese type 2 diabetes rats. Bone 2008, 42, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Rharass, T.; Lucas, S. High Glucose Level Impairs Human Mature Bone Marrow Adipocyte Function Through Increased ROS Production. Front. Endocrinol. 2019, 10, 607. [Google Scholar] [CrossRef] [PubMed]
- Ko, K.I.; Syverson, A.L.; Kralik, R.M.; Choi, J.; DerGarabedian, B.P.; Chen, C.; Graves, D.T. Diabetes-Induced NF-kappaB Dysregulation in Skeletal Stem Cells Prevents Resolution of Inflammation. Diabetes 2019, 68, 2095–2106. [Google Scholar] [CrossRef] [Green Version]
- Halade, G.V.; El Jamali, A.; Williams, P.J.; Fajardo, R.J.; Fernandes, G. Obesity-mediated inflammatory microenvironment stimulates osteoclastogenesis and bone loss in mice. Exp. Gerontol. 2011, 46, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Kayal, R.A.; Siqueira, M.; Alblowi, J.; McLean, J.; Krothapalli, N.; Faibish, D.; Einhorn, T.A.; Gerstenfeld, L.C.; Graves, D.T. TNF-alpha mediates diabetes-enhanced chondrocyte apoptosis during fracture healing and stimulates chondrocyte apoptosis through FOXO1. J. Bone Min. Res. 2010, 25, 1604–1615. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.C.; Ko, K.I.; Mattos, M.; Fang, M.; Zhang, C.; Feinberg, D.; Sindi, H.; Li, S.; Alblowi, J.; Kayal, R.A.; et al. TNFalpha contributes to diabetes impaired angiogenesis in fracture healing. Bone 2017, 9926–9938. [Google Scholar] [CrossRef]
- Cassidy, F.C.; Shortiss, C.; Murphy, C.G.; Kearns, S.R.; Curtin, W.; De Buitleir, C.; O’Brien, T.; Coleman, C.M. Impact of Type 2 Diabetes Mellitus on Human Bone Marrow Stromal Cell Number and Phenotypic Characteristics. Int. J. Mol. Sci. 2020, 21, 2476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aliakbari, S.; Mohammadi, M.; Rezaee, M.A.; Amini, A.A.; Fakhari, S.; Rahmani, M.R. Impaired immunomodulatory ability of type 2 diabetic adipose-derived mesenchymal stem cells in regulation of inflammatory condition in mixed leukocyte reaction. Excli. J. 2019, 18852–18865. [Google Scholar] [CrossRef]
- Filion, T.M.; Skelly, J.D.; Huang, H.; Greiner, D.L.; Ayers, D.C.; Song, J. Impaired osteogenesis of T1DM bone marrow-derived stromal cells and periosteum-derived cells and their differential in-vitro responses to growth factor rescue. Stem Cell Res. Ther. 2017, 8, 65. [Google Scholar] [CrossRef] [Green Version]
- Wallner, C.; Schira, J.; Wagner, J.M.; Schulte, M.; Fischer, S.; Hirsch, T.; Richter, W.; Abraham, S.; Kneser, U.; Lehnhardt, M.; et al. Application of VEGFA and FGF-9 enhances angiogenesis, osteogenesis and bone remodeling in type 2 diabetic long bone regeneration. PLoS ONE 2015, 10, e0118823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.M.; Tao, J.; Chen, D.D.; Cai, J.J.; Irani, K.; Wang, Q.; Yuan, H.; Chen, A.F. MicroRNA miR-27b rescues bone marrow-derived angiogenic cell function and accelerates wound healing in type 2 diabetes mellitus. Arter. Thromb. Vasc. Biol. 2014, 34, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Bae, O.N.; Wang, J.M.; Baek, S.H.; Wang, Q.; Yuan, H.; Chen, A.F. Oxidative stress-mediated thrombospondin-2 upregulation impairs bone marrow-derived angiogenic cell function in diabetes mellitus. Arter. Thromb. Vasc. Biol. 2013, 33, 1920–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.H.; Park, H.K.; Choi, N.R.; Kim, S.W.; Kim, G.C.; Hwang, D.S.; Kim, Y.D.; Shin, S.H.; Kim, U.K. Relationship between disease stage and renal function in bisphosphonate-related osteonecrosis of the jaw. J. Korean Assoc. Oral Maxillofac. Surg. 2017, 43, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Boechat, M.I.; Winters, W.D.; Hogg, R.J.; Fine, R.N.; Watkins, S.L. Avascular necrosis of the femoral head in children with chronic renal disease. Radiology 2001, 218, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Abbott, K.C.; Oglesby, R.J.; Agodoa, L.Y. Hospitalized avascular necrosis after renal transplantation in the United States. Kidney Int. 2002, 62, 2250–2256. [Google Scholar] [CrossRef] [Green Version]
- Klinkhammer, B.M.; Kramann, R.; Mallau, M.; Makowska, A.; van Roeyen, C.R.; Rong, S.; Buecher, E.B.; Boor, P.; Kovacova, K.; Zok, S.; et al. Mesenchymal stem cells from rats with chronic kidney disease exhibit premature senescence and loss of regenerative potential. PLoS ONE 2014, 9, e92115. [Google Scholar] [CrossRef] [Green Version]
- Gangji, V.; Hauzeur, J.P.; Matos, C.; De Maertelaer, V.; Toungouz, M.; Lambermont, M. Treatment of osteonecrosis of the femoral head with implantation of autologous bone-marrow cells. A pilot study. J. Bone Jt. Surg. Am. 2004, 86, 1153–1160. [Google Scholar] [CrossRef] [Green Version]
- Kawate, K.; Yajima, H.; Ohgushi, H.; Kotobuki, N.; Sugimoto, K.; Ohmura, T.; Kobata, Y.; Shigematsu, K.; Kawamura, K.; Tamai, K.; et al. Tissue-engineered approach for the treatment of steroid-induced osteonecrosis of the femoral head: Transplantation of autologous mesenchymal stem cells cultured with beta-tricalcium phosphate ceramics and free vascularized fibula. Artif Organs 2006, 30, 960–962. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.L.; Sun, W.; Shi, Z.C.; Zhang, N.F.; Yue, D.B.; Guo, W.S.; Xu, S.Q.; Lou, J.N.; Li, Z.R. Treatment of nontraumatic osteonecrosis of the femoral head with the implantation of core decompression and concentrated autologous bone marrow containing mononuclear cells. Arch. Orthop. Trauma Surg. 2010, 130, 859–865. [Google Scholar] [CrossRef]
- Mao, Q.; Jin, H.; Liao, F.; Xiao, L.; Chen, D.; Tong, P. The efficacy of targeted intraarterial delivery of concentrated autologous bone marrow containing mononuclear cells in the treatment of osteonecrosis of the femoral head: A five year follow-up study. Bone 2013, 57, 509–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, K.; MacKenzie, M.J.; Winquist, E. Chemotherapy-associated osteonecrosis in cancer patients with solid tumours: A systematic review. Drug Saf. 2008, 31, 359–371. [Google Scholar] [CrossRef]
- Somaiah, C.; Kumar, A.; Sharma, R.; Sharma, A.; Anand, T.; Bhattacharyya, J.; Das, D.; Deka Talukdar, S.; Jaganathan, B.G. Mesenchymal stem cells show functional defect and decreased anti-cancer effect after exposure to chemotherapeutic drugs. J. Biomed. Sci. 2018, 25, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Law, H.K.; Lau, Y.L.; Chan, G.C. Differential damage and recovery of human mesenchymal stem cells after exposure to chemotherapeutic agents. Br. J. Haematol. 2004, 127, 326–334. [Google Scholar] [CrossRef]
- Nicolay, N.H.; Ruhle, A.; Perez, R.L.; Trinh, T.; Sisombath, S.; Weber, K.J.; Ho, A.D.; Debus, J.; Saffrich, R.; Huber, P.E. Mesenchymal stem cells are sensitive to bleomycin treatment. Sci. Rep. 2016, 626645. [Google Scholar] [CrossRef] [PubMed]
- Nifontova, I.; Svinareva, D.; Petrova, T.; Drize, N. Sensitivity of mesenchymal stem cells and their progeny to medicines used for the treatment of hematoproliferative diseases. Acta Haematol. 2008, 119, 98–103. [Google Scholar] [CrossRef]
- Oliveira, M.S.; Carvalho, J.L.; Campos, A.C.; Gomes, D.A.; de Goes, A.M.; Melo, M.M. Doxorubicin has in vivo toxicological effects on ex vivo cultured mesenchymal stem cells. Toxicol. Lett. 2014, 224, 380–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galotto, M.; Berisso, G.; Delfino, L.; Podesta, M.; Ottaggio, L.; Dallorso, S.; Dufour, C.; Ferrara, G.B.; Abbondandolo, A.; Dini, G.; et al. Stromal damage as consequence of high-dose chemo/radiotherapy in bone marrow transplant recipients. Exp. Hematol. 1999, 27, 1460–1466. [Google Scholar] [CrossRef]
- Kemp, K.; Morse, R.; Wexler, S.; Cox, C.; Mallam, E.; Hows, J.; Donaldson, C. Chemotherapy-induced mesenchymal stem cell damage in patients with hematological malignancy. Ann. Hematol. 2010, 89, 701–713. [Google Scholar] [CrossRef] [Green Version]
- Prata Kde, L.; Orellana, M.D.; De Santis, G.C.; Kashima, S.; Fontes, A.M.; Carrara Rde, C.; Palma, P.V.; Neder, L.; Covas, D.T. Effects of high-dose chemotherapy on bone marrow multipotent mesenchymal stromal cells isolated from lymphoma patients. Exp. Hematol. 2010, 38, 292–300.e4. [Google Scholar] [CrossRef]
- Li, R.; Lin, Q.X.; Liang, X.Z.; Liu, G.B.; Tang, H.; Wang, Y.; Lu, S.B.; Peng, J. Stem cell therapy for treating osteonecrosis of the femoral head: From clinical applications to related basic research. Stem Cell Res. Ther. 2018, 9, 291. [Google Scholar] [CrossRef] [PubMed]
- Kraft, D.L.; Walck, E.R.; Carrasco, A.; Crocker, M.D.; Song, L.; Long, M.G.; Mosse, M.A.; Nadeem, B.; Imanbayev, G.T.; Czechowicz, A.D.; et al. The MarrowMiner: A Novel Minimally Invasive and Effective Device for the Harvest of Bone Marrow. Biol. Blood Marrow Transpl. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Z.M.; Jiang, H.; Zhan, X.L.; Wu, Z.G.; Zhang, X.L. Treatment of osteonecrosis of femoral head with BMSCs-seeded bio-derived bone materials combined with rhBMP-2 in rabbits. Chin. J. Traumatol. 2008, 11, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.T.; Lu, B.; Yue, B.; Xie, X.H.; Xie, Y.Z.; Dai, K.R.; Lu, J.X.; Lou, J.R. Treatment of osteonecrosis of the femoral head with hBMP-2-gene-modified tissue-engineered bone in goats. J. Bone Jt. Surg. Br. 2007, 89, 127–129. [Google Scholar] [CrossRef] [Green Version]
- Wen, Q.; Ma, L.; Chen, Y.P.; Yang, L.; Luo, W.; Wang, X.N. Treatment of avascular necrosis of the femoral head by hepatocyte growth factor-transgenic bone marrow stromal stem cells. Gene Ther. 2008, 15, 1523–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, C.S.; Yesantharao, P.; Huang, C.Y.; Mattson, G.; Boktor, J.; Fukunishi, T.; Zhang, H.; Hibino, N. 3D bioprinting using stem cells. Pediatric Res. 2018, 83, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Wu, X.; Chen, X.; Zhou, P.; Xu, F.; Liang, W. Nanotechnology shaping stem cell therapy: Recent advances, application, challenges, and future outlook. Biomed. Pharm. 2021, 137, 111236. [Google Scholar] [CrossRef]
- El-Jawhari, J.J.; Ilas, D.C.; Jones, W.; Cuthbert, R.; Jones, E.; Giannoudis, P.V. Enrichment and preserved functionality of multipotential stromal cells in bone marrow concentrate processed by vertical centrifugation. Eur. Cell Mater. 2020, 4058–4073. [Google Scholar] [CrossRef]
- El-Jawhari, J.J.; Ganguly, P.; Churchman, S.; Jones, E.; Giannoudis, P.V. The Biological Fitness of Bone Progenitor Cells in Reamer/Irrigator/Aspirator Waste. J. Bone Jt. Surg. Am. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Qu, Z.; Yin, X.; Shang, C.; Ao, Q.; Gu, Y.; Liu, Y. Efficacy of umbilical cord-derived mesenchymal stem cell-based therapy for osteonecrosis of the femoral head: A three-year follow-up study. Mol. Med. Rep. 2016, 14, 4209–4215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, D.; Kumar, S.; Webster, T.J.; Ramalingam, M. Impact of Induced Pluripotent Stem Cells in Bone Repair and Regeneration. Curr. Osteoporos. Rep. 2019, 17, 226–234. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Age (Years) | Volume (mL) | Isolation | Media | Colony Definition | CFU-F | Ref |
|---|---|---|---|---|---|---|
| Y: 22–44, O: 66–74 | 10 mL | DC | α-MEM + 10%FCS | >16 cells | No change | [67] |
| 13–79, no groups | 4 × 2 mL pooled | DC | α-MEM + 10%FCS + ASC + Dex | >8 cells | No change | [68] |
| Y: 0–18, O: 59–75 | NR | DC | DMEM + 10%FCS | NR | Decline | [58] |
| Y: 19–40, O: > 40 | NR | DC | DMEM + 10%FCS | >50 cells | Decline | [69] |
| Y: 6–16, O: 29–76 | NR | PA | DMEM + 20%FCS | NR | NS decline | [70] |
| 1–52, no groups | NR | DC | DMEM + 20%FCS | >50 cells | No change | [71] |
| 22–80, no groups | 8 mL | PA | StemMacs medium | >50 cells | Decline in women | [72] |
| Y: 20–40, I: 41–60, O: >60 | 10 mL | PA | StemMacs medium | >50 cells | Decline | [73] |
| 14–59, no groups | 30 mL, 3 × 10 mL | PA | DMEM/Ham’s F12 + 10% FCS + bFGF + heparin | >50 cells | Decline | [74] |
| Y: <45, I: 45–65, O: >65 | NR | DC | α-MEM + 10% human serum | >50 cells | Decline | [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Jawhari, J.J.; Ganguly, P.; Jones, E.; Giannoudis, P.V. Bone Marrow Multipotent Mesenchymal Stromal Cells as Autologous Therapy for Osteonecrosis: Effects of Age and Underlying Causes. Bioengineering 2021, 8, 69. https://doi.org/10.3390/bioengineering8050069
El-Jawhari JJ, Ganguly P, Jones E, Giannoudis PV. Bone Marrow Multipotent Mesenchymal Stromal Cells as Autologous Therapy for Osteonecrosis: Effects of Age and Underlying Causes. Bioengineering. 2021; 8(5):69. https://doi.org/10.3390/bioengineering8050069
Chicago/Turabian StyleEl-Jawhari, Jehan J, Payal Ganguly, Elena Jones, and Peter V Giannoudis. 2021. "Bone Marrow Multipotent Mesenchymal Stromal Cells as Autologous Therapy for Osteonecrosis: Effects of Age and Underlying Causes" Bioengineering 8, no. 5: 69. https://doi.org/10.3390/bioengineering8050069
APA StyleEl-Jawhari, J. J., Ganguly, P., Jones, E., & Giannoudis, P. V. (2021). Bone Marrow Multipotent Mesenchymal Stromal Cells as Autologous Therapy for Osteonecrosis: Effects of Age and Underlying Causes. Bioengineering, 8(5), 69. https://doi.org/10.3390/bioengineering8050069