
Pan-Genome Analysis Reveals Functional Divergences in Gut-Restricted Gilliamella and Snodgrassella

Abstract
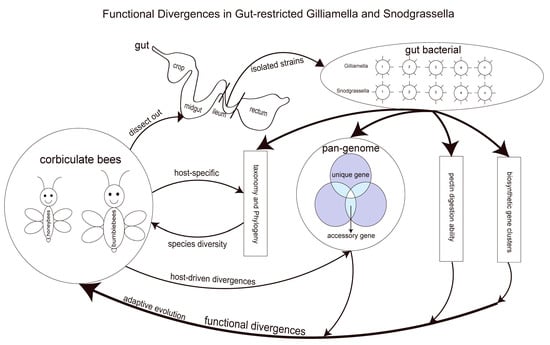
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Bee Sample Collection, Culture, and Identification of Gut Bacteria
2.2. Isolates DNA Extraction, Library Preparation, and Sequencing
2.3. Genomic Data Collection and Bioinformatics Analysis
3. Results
3.1. Population Delimitation within Gilliamella Genus and Snodgrassella Genus Based on ANI and GTDB Analysis
3.2. Phylogeny Reconstruction for Gilliamella Genus and Snodgrassella Genus
3.3. Pan-Genome Analysis of Gilliamella and Snodgrassella
3.4. Pan-Genome Analysis of Gilliamella and Snodgrassella with Different Genus Hosts
3.5. Distribution of Genes Related to Carbohydrate Metabolism
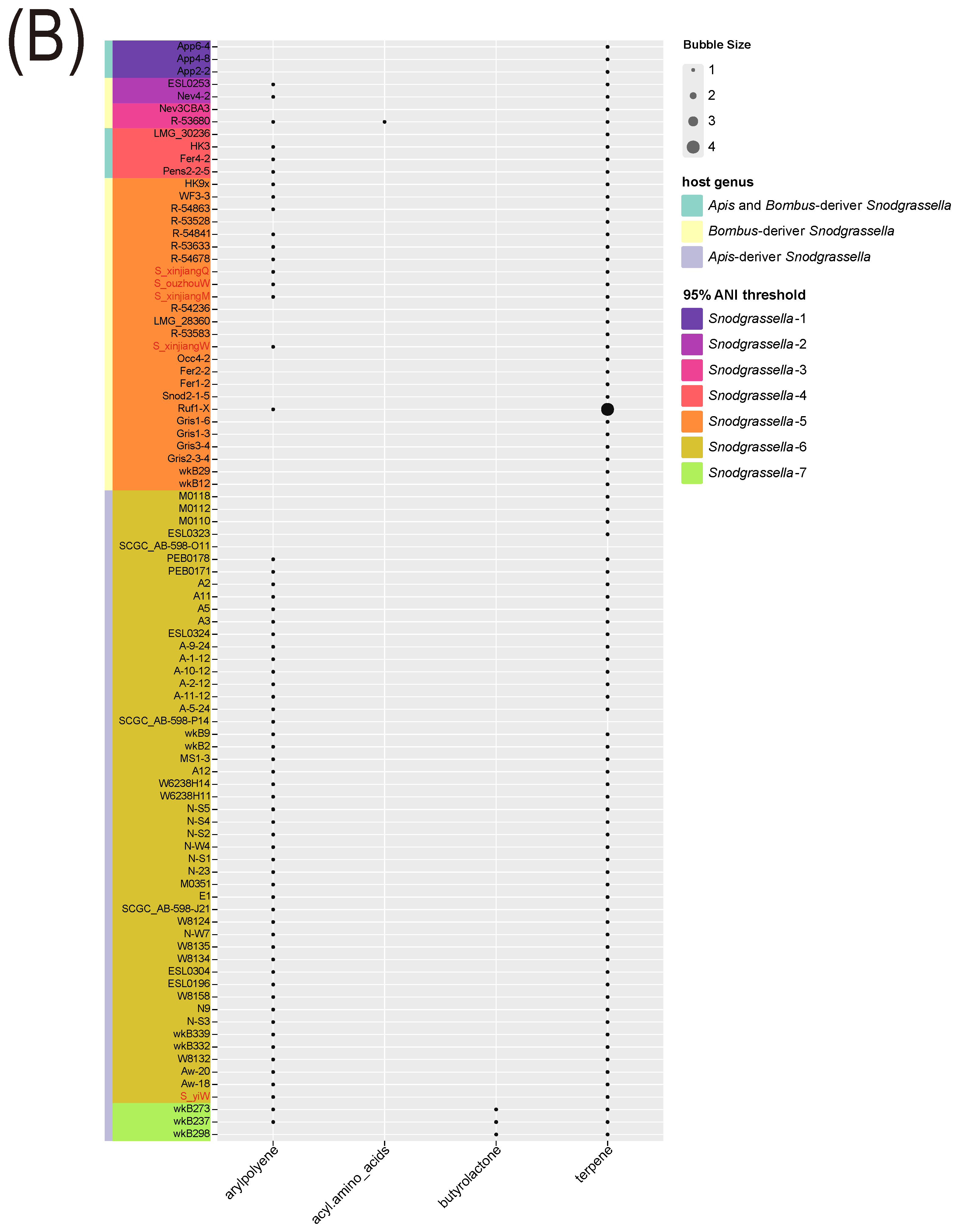
3.6. Secondary Metabolite Analysis for Gilliamella Genus and Snodgrassella Genus
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engel, M.S.; Rasmussen, C. Corbiculate Bees. In Encyclopedia of Social Insects; Starr, C.K., Ed.; Springer: Cham, Switzerland, 2021. [Google Scholar] [CrossRef]
- Stein, K.; Coulibaly, D.; Stenchly, K.; Goetze, D.; Porembski, S.; Lindner, A.; Konate, S.; Linsenmair, E.K. Bee pollination increases yield quantity and quality of cash crops in Burkina Faso, West Africa. Sci. Rep. 2017, 7, 17691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraemer, S.A.; Ramachandran, A.; Perron, G.G. Antibiotic Pollution in the Environment: From Microbial Ecology to Public Policy. Microorganisms 2019, 7, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, W.K.; Moran, N.A. Gut microbial communities of social bees. Nat. Rev. Microbiol. 2016, 14, 374–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, X.; Zhang, Z.; Lang, H.; Zheng, H. Honey bee as a model organism to study gut microbiota and diseases. Drug Discov. Today Dis. Models 2018, 28, 35–42. [Google Scholar] [CrossRef]
- Zheng, H.; Perreau, J.; Powell, J.E.; Han, B.; Zhang, Z.; Kwong, W.K.; Tringe, S.G.; Moran, N.A. Division of labor in honey bee gut microbiota for plant polysaccharide digestion. Proc. Natl. Acad. Sci. USA 2019, 116, 25909–25916. [Google Scholar] [CrossRef]
- Horak, R.D.; Leonard, S.P.; Moran, N.A. Symbionts shape host innate immunity in honeybees. Proc. R. Soc. B 2020, 287, 20201184. [Google Scholar] [CrossRef]
- Kwong, W.K.; Moran, N.A. Cultivation and characterization of the gut symbionts of honey bees and bumble bees: Description of Snodgrassella alvi gen. nov., sp. nov., a member of the family Neisseriaceae of the Betaproteobacteria, and Gilliamella apicola gen. nov., sp. nov., a member of Orbaceae fam. nov., Orbales ord. nov., a sister taxon to the order ‘Enterobacteriales’ of the Gammaproteobacteria. Int. J. Syst. Evol. Microbiol. 2013, 63, 2008–2018. [Google Scholar] [CrossRef]
- Janda, J.M.; Abbott, S.L. 16S rRNA Gene Sequencing for Bacterial Identification in the Diagnostic Laboratory: Pluses, Perils, and Pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. nt. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez, R.L.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornet, L.; Cleenwerck, I.; Praet, J.; Leonard, R.R.; Vereecken, N.J.; Michez, D.; Smagghe, G.; Baurain, D.; Vandamme, P. Phylogenomic Analyses of Snodgrassella Isolates from Honeybees and Bumblebees Reveal Taxonomic and Functional Diversity. mSystems 2022, 7, e0150021. [Google Scholar] [CrossRef] [PubMed]
- Ptaszynska, A.A.; Latoch, P.; Hurd, P.J.; Polaszek, A.; Michalska-Madej, J.; Grochowalski, L.; Strapagiel, D.; Gnat, S.; Zaluski, D.; Gancarz, M.; et al. Amplicon Sequencing of Variable 16S rRNA from Bacteria and ITS2 Regions from Fungi and Plants, Reveals Honeybee Susceptibility to Diseases Results from Their Forage Availability under Anthropogenic Landscapes. Pathogens 2021, 10, 381. [Google Scholar] [CrossRef] [PubMed]
- Kwong, W.K.; Medina, L.A.; Koch, H.; Sing, K.W.; Soh, E.J.Y.; Ascher, J.S.; Jaffé, R.; Moran, N.A. Dynamic microbiome evolution in social bees. Sci. Adv. 2017, 3, e1600513. [Google Scholar] [CrossRef] [Green Version]
- Kwong, W.K.; Engel, P.; Koch, H.; Moran, N.A. Genomics and host specialization of honey bee and bumble bee gut symbionts. Proc. Natl. Acad. Sci. USA 2014, 111, 11509–11514. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Kitamoto, S.; Kamada, N. Microbial adaptation to the healthy and inflamed gut environments. Gut Microbes 2020, 12, 1857505. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Huang, M.F.; Qiu, L.F.; Song, R.H.; Zhang, Z.X.; Ding, Y.W.; Zhou, X.; Zhang, X.; Zheng, H. Diversity and functional analysis of Chinese bumblebee gut microbiota reveal the metabolic niche and antibiotic resistance variation of Gilliamella. Insect Sci. 2021, 28, 302–314. [Google Scholar] [CrossRef]
- Li, Y.; Leonard, S.P.; Powell, J.E.; Moran, N.A. Species divergence in gut-restricted bacteria of social bees. Proc. Natl. Acad. Sci. USA 2022, 119, e2115013119. [Google Scholar] [CrossRef]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef]
- Sherman, R.M.; Salzberg, S.L. Pan-genomics in the human genome era. Nat. Rev. Genet. 2020, 21, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Paulos, S.; Mateo, M.; de Lucio, A.; Hernandez-de Mingo, M.; Bailo, B.; Saugar, J.M.; Cardona, G.A.; Fuentes, I.; Mateo, M.; Carmena, D. Evaluation of five commercial methods for the extraction and purification of DNA from human faecal samples for downstream molecular detection of the enteric protozoan parasites Cryptosporidium spp., Giardia duodenalis, and Entamoeba spp. J. Microbiol. Methods 2016, 127, 68–73. [Google Scholar] [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef] [PubMed]
- Alexey, G.; Vladislav, S.; Nikolay, V.; Glenn, T. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Carroll, L.M.; Wiedmann, M.; Kovac, J. Proposal of a Taxonomic Nomenclature for the Bacillus cereus Group Which Reconciles Genomic Definitions of Bacterial Species with Clinical and Industrial Phenotypes. mBio 2020, 11, e00034-20. [Google Scholar] [CrossRef] [Green Version]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Busk, P.K.; Pilgaard, B.; Lezyk, M.J.; Meyer, A.S.; Lange, L. Homology to peptide pattern for annotation of carbohydrate-active enzymes and prediction of function. BMC Bioinform. 2017, 18, 214. [Google Scholar] [CrossRef] [Green Version]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [Green Version]
- Praet, J.; Cnockaert, M.; Meeus, I.; Smagghe, G.; Vandamme, P. Gilliamella intestini sp. nov., Gilliamella bombicola sp. nov., Gilliamella bombi sp. nov. and Gilliamella mensalis sp. nov.: Four novel Gilliamella species isolated from the bumblebee gut. Syst. Appl. Microbiol. 2017, 40, 199–204. [Google Scholar] [CrossRef]
- Rouli, L.; Merhej, V.; Fournier, P.E.; Raoult, D. The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect. 2015, 7, 72–85. [Google Scholar] [CrossRef]
- Moran, N.A.; Ochman, H.; Hammer, T.J. Evolutionary and ecological consequences of gut microbial communities. Annu. Rev. Ecol. Evol. Syst. 2019, 50, 451–475. [Google Scholar] [CrossRef] [PubMed]
- Marroni, F.; Pinosio, S.; Morgante, M. Structural variation and genome complexity: Is dispensable really dispensable? Curr. Opin. Plant Biol. 2014, 18, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Park, J.; Jung, H.; Seo, Y.S. Pan-Genome Analysis Reveals Host-Specific Functional Divergences in Burkholderia gladioli. Microorganisms 2021, 9, 1123. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.E.; Leonard, S.P.; Kwong, W.K.; Engel, P.; Moran, N.A. Genome-wide screen identifies host colonization determinants in a bacterial gut symbiont. Proc. Natl. Acad. Sci. USA 2016, 113, 13887–13892. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.A.; Macfarlane, G.T. Dissimilatory Amino Acid Metabolism in Human Colonic Bacteria. Anaerobe 1997, 3, 327–337. [Google Scholar] [CrossRef]
- Richardson, A.D.; Moscow, J.A. Can an enzyme cofactor be a factor in malignant progression? Cancer Biol. Ther. 2010, 10, 1112–1114. [Google Scholar] [CrossRef] [Green Version]
- Bender, D.A.; Mayes, P.A. The Citric Acid Cycle: The Central Pathway of Carbohydrate, Lipid & Amino Acid Metabolism. In Harper’s Illustrated Biochemistry, 30e; Rodwell, V.W., Bender, D.A., Botham, K.M., Kennelly, P.J., Weil, P.A., Eds.; McGraw-Hill Education: New York, NY, USA, 2016. [Google Scholar]
- Vanderlinde, E.M.; Harrison, J.J.; Muszynski, A.; Carlson, R.W.; Turner, R.J.; Yost, C.K. Identification of a novel ABC transporter required for desiccation tolerance, and biofilm formation in Rhizobium leguminosarum bv. viciae 3841. EMS Microbiol. Ecol. 2010, 71, 327–340. [Google Scholar] [CrossRef] [Green Version]
- Schoner, T.A.; Gassel, S.; Osawa, A.; Tobias, N.J.; Okuno, Y.; Sakakibara, Y.; Shindo, K.; Sandmann, G.; Bode, H.B. Aryl Polyenes, a Highly Abundant Class of Bacterial Natural Products, Are Functionally Related to Antioxidative Carotenoids. Chembiochem 2016, 17, 247–253. [Google Scholar] [CrossRef]
- Jones, R.M.; Mercante, J.W.; Neish, A.S. Reactive Oxygen Production Induced by the Gut Microbiota: Pharmacotherapeutic Implications. Curr. Med. Chem. 2012, 19, 1519–1529. [Google Scholar] [CrossRef]
- Aviello, G.; Knaus, U.G. ROS in gastrointestinal inflammation: Rescue Or Sabotage? Br. J. Pharmacol. 2017, 174, 1704–1718. [Google Scholar] [CrossRef]
- Burenina, O.Y.; Elkina, D.A.; Ovcharenko, A.; Bannikova, V.A.; Schluter, M.A.C.; Oretskaya, T.S.; Hartmann, R.K.; Kubareva, E.A. Involvement of E. coli 6S RNA in Oxidative Stress Response. Int. J. Mol. Sci. 2022, 23, 3653. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, W.; Li, J.; Li, Y.; Zhang, J.; Tan, H. A widespread response of Gram-negative bacterial acyl-homoserine lactone receptors to Gram-positive Streptomyces gamma-butyrolactone signaling molecules. Sci. China Life Sci. 2021, 64, 1575–1589. [Google Scholar] [CrossRef] [PubMed]
- Peyrottes, A. Modulation of Inflammation in Intestinal Epithelial and Immune Cells by N-Acyl Homoserine Lactones and their Synthetic Analogues. Ph.D. Thesis, Sorbonne Université, Paris, France, 2019. [Google Scholar]
- Just-Baringo, X.; Albericio, F.; Alvarez, M. Thiopeptide antibiotics: Retrospective and recent advances. Mar Drugs 2014, 12, 317–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleich, R.; Watrous, J.D.; Dorrestein, P.C.; Bowers, A.A.; Shank, E.A. Thiopeptide antibiotics stimulate biofilm formation in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2015, 112, 3086–3091. [Google Scholar] [CrossRef] [Green Version]
- Felnagle, E.A.; Jackson, E.E.; Chan, Y.A.; Podevels, A.M.; Berti, A.D.; McMahon, M.D.; Thomas, M.G. Nonribosomal peptide synthetases involved in the production of medically relevant natural products. Mol. Pharm. 2008, 5, 191–211. [Google Scholar] [CrossRef] [PubMed]
- Ronnebaum, T.A.; Lamb, A.L. Nonribosomal peptides for iron acquisition: Pyochelin biosynthesis as a case study. Curr. Opin. Struct. Biol. 2018, 53, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zengin, H.; Baysal, A.H. Antibacterial and antioxidant activity of essential oil terpenes against pathogenic and spoilage-forming bacteria and cell structure-activity relationships evaluated by SEM microscopy. Molecules 2014, 19, 17773–17798. [Google Scholar] [CrossRef] [Green Version]
- Downer, E.J. Anti-inflammatory Potential of Terpenes Present in Cannabis sativa L. ACS Chem. Neurosci. 2020, 11, 659–662. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Guo, Y.; Yang, F.; Li, J. Pan-Genome Analysis Reveals Functional Divergences in Gut-Restricted Gilliamella and Snodgrassella. Bioengineering 2022, 9, 544. https://doi.org/10.3390/bioengineering9100544
Zhang Z, Guo Y, Yang F, Li J. Pan-Genome Analysis Reveals Functional Divergences in Gut-Restricted Gilliamella and Snodgrassella. Bioengineering. 2022; 9(10):544. https://doi.org/10.3390/bioengineering9100544
Chicago/Turabian StyleZhang, Zhengyi, Yulong Guo, Fan Yang, and Jilian Li. 2022. "Pan-Genome Analysis Reveals Functional Divergences in Gut-Restricted Gilliamella and Snodgrassella" Bioengineering 9, no. 10: 544. https://doi.org/10.3390/bioengineering9100544
APA StyleZhang, Z., Guo, Y., Yang, F., & Li, J. (2022). Pan-Genome Analysis Reveals Functional Divergences in Gut-Restricted Gilliamella and Snodgrassella. Bioengineering, 9(10), 544. https://doi.org/10.3390/bioengineering9100544