Abstract
Prostate cancer (PCa) is a major health burden worldwide, and despite early treatment, many patients present with biochemical recurrence (BCR) post-treatment, reflected by a rise in prostate-specific antigen (PSA) over a clinical threshold. Novel transcriptomic and epigenomic biomarkers can provide a powerful tools for the clinical management of PCa. Here, we provide matched RNA sequencing and array-based genome-wide DNA methylome data of PCa patients (n = 17) with or without evidence of BCR following radical prostatectomy. Formalin-fixed paraffin-embedded (FFPE) tissues were used to generate these data, which included technical replicates to provide further validity of the data. We describe the sample features, experimental design, methods and bioinformatic pipelines for processing these multi-omic data. Importantly, comprehensive clinical, histopathological, and follow-up data for each patient were provided to enable the correlation of transcriptome and methylome features with clinical features. Our data will contribute towards the efforts of developing epigenomic and transcriptomic markers for BCR and also facilitate a deeper understanding of the molecular basis of PCa recurrence.
Dataset: MethylationEPIC v2.0 array and RNA-Seq data for this study can be found in the GEO database under accession number GSE282574.
Dataset Licence: Licence under which the dataset is made available: CC BY-NC-ND.
1. Background and Summary
Prostate cancer (PCa) is the second most commonly diagnosed malignancy in males worldwide, accounting for 7.3% of new cancer diagnoses and almost 400,000 deaths annually [1]. For men with clinically significant localised disease, curative intent treatment with radical prostatectomy or radiation therapy is the standard of care. Despite early definitive treatment, however, 10–60% of patients will develop biochemical recurrence (BCR) of PCa within 10 years [2,3]. In the context of definitive surgical management, BCR is defined as a persisting rise in prostate-specific antigen (PSA) level to ≥0.20 ng/mL after radical prostatectomy [4,5]. The clinical significance of BCR can be challenging to interpret, with PSA monitoring alone offering limited prognostic value in determining the expected clinical progression in an individual patient [6]. Nevertheless, for patients with specific clinical risk factors, including short PSA doubling time and high Gleason score at prostatectomy, BCR increases prostate cancer-specific mortality rates and the risk of metastatic progression [7]. Efforts to develop more accurate risk stratification tools in the context of BCR have been undertaken; however, long-term clinical outcomes remain difficult to predict accurately, which presents challenges for clinical decision-making and appropriate initiation of salvage therapies [8,9].
Novel molecular biomarkers, as well as improved knowledge of the key pathobiological pathways implicated in disease recurrence post-prostatectomy, represent a potential solution. Consensus genomic alterations are scarce in localised PCa, and in this context, epigenomic variations may herald greater prospective clinical utility [10]. Epigenetic changes, specifically DNA methylation alterations, are a critical factor in tumour development, progression, and metastasis in multiple cancer types [11,12,13,14]. Indeed, dynamic epigenetic alteration of DNA methylation profiles and subsequent transcriptomic dysregulation has now been characterised across all stages of PCa [15,16,17,18,19]. Further, for less invasive cell-free DNA sampling, driver epigenetic alterations are cancer-type specific and much higher in number than genetic mutations, thus enabling earlier tumour detection. There is growing interest in the integration of DNA methylation signatures to enhance PCa detection, prognostication, and disease monitoring, although further research is required to facilitate successful clinical translation.
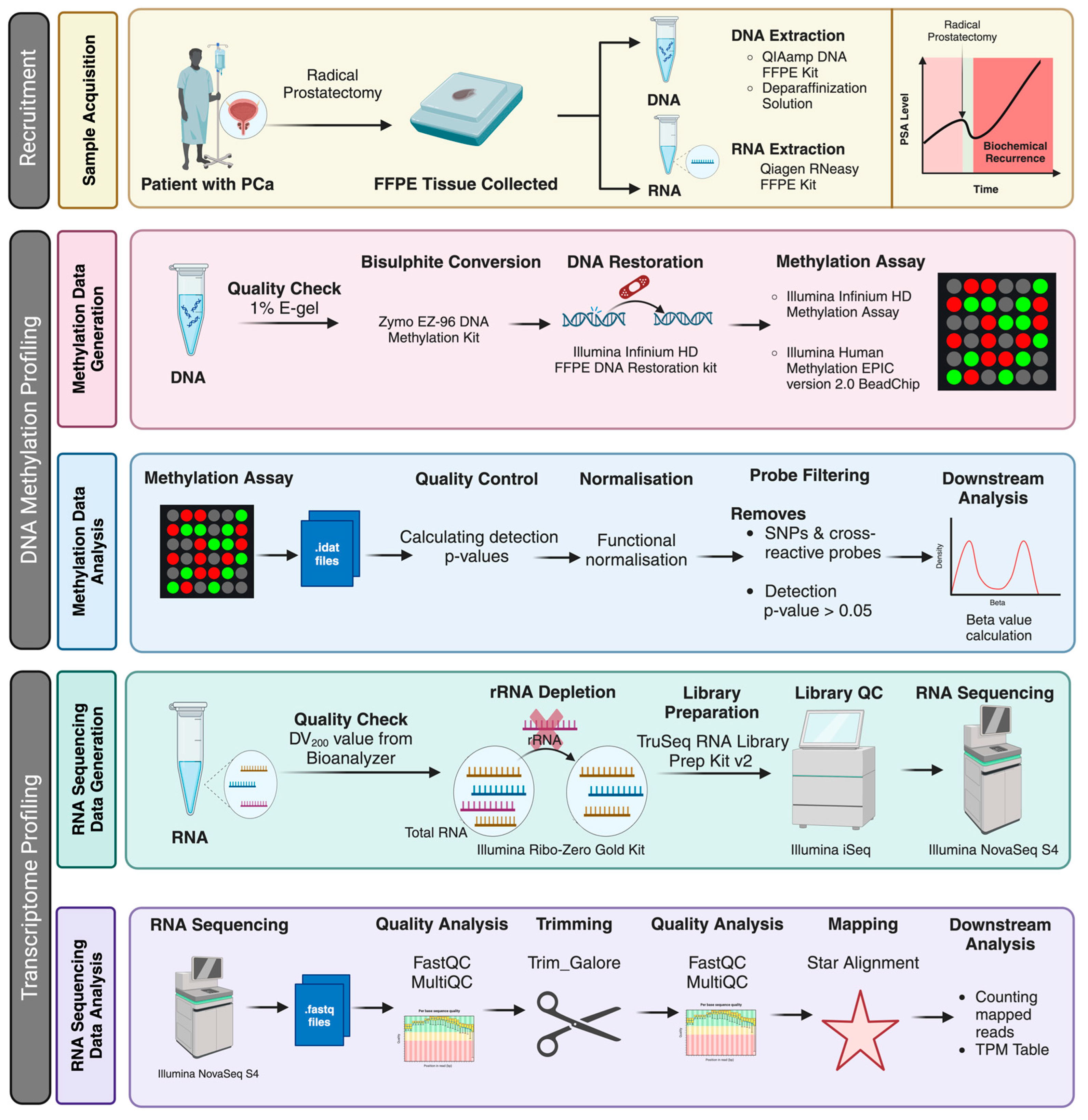
Here, we contribute DNA methylomic and transcriptional data in PCa, with a specific focus on characterising the molecular profiles of BCR post-prostatectomy. We have generated matched, genome-scale profiles of the DNA methylome (MethylationEPIC v2.0 array) and RNA transcriptome (RNA-Seq) for a cohort of 17 radical prostatectomy patients with replicates to assess reproducibility. We provide a detailed workflow for processing these tissue samples, molecular data generation, and subsequent bioinformatics analysis pipelines (Figure 1). Comprehensive clinical and histopathological data are available for all participants, allowing stratification of this cohort into those with and without evidence of BCR within four years following prostatectomy.
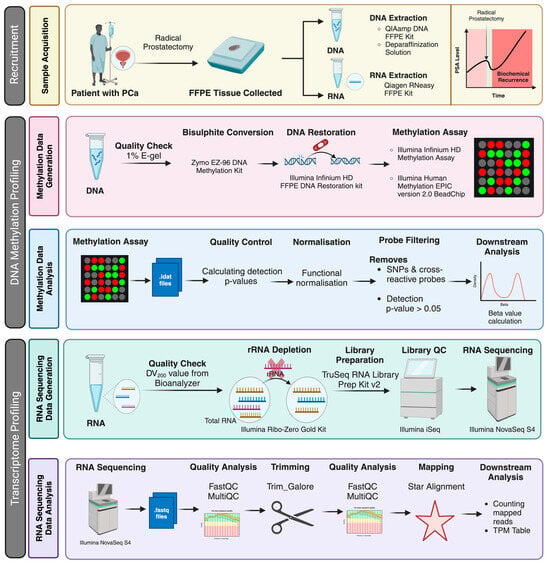
Figure 1.
Schematic representation of the experiment workflow. The figure describes an overview of the patient recruitment (the status of biochemical recurrence of the patients was retrieved later for grouping the patients), analysis strategy and platforms used to generate the data described in this work. Created using BioRender.com. https://www.biorender.com (accessed on 8 August 2024).
2. Data Description
Patient data, including relevant demographic, histopathological, and clinical data, were obtained from medical records, as described in the Methods section. Key data for each respective patient sample are detailed in Table 1 alongside their respective MethylationEPIC v2.0 array and RNA-Seq accession numbers. Additional patient information and clinical data for each patient sample are provided in Supplementary Table S1, including detailed pathology report information for both prostate biopsy and radical prostatectomy samples from each participant. All MethylationEPIC v2.0 array and RNA-Seq data for this study can be found in the GEO database under accession number GSE282574. Each data file is assigned an accession number and has a ‘GSM’ prefix (Table 1).

Table 1.
Individual sample information with MethylationEPIC v2.0 and RNA sequencing accession numbers.
3. Results
3.1. DNA Methylome Data Analysis, Assessments and Features
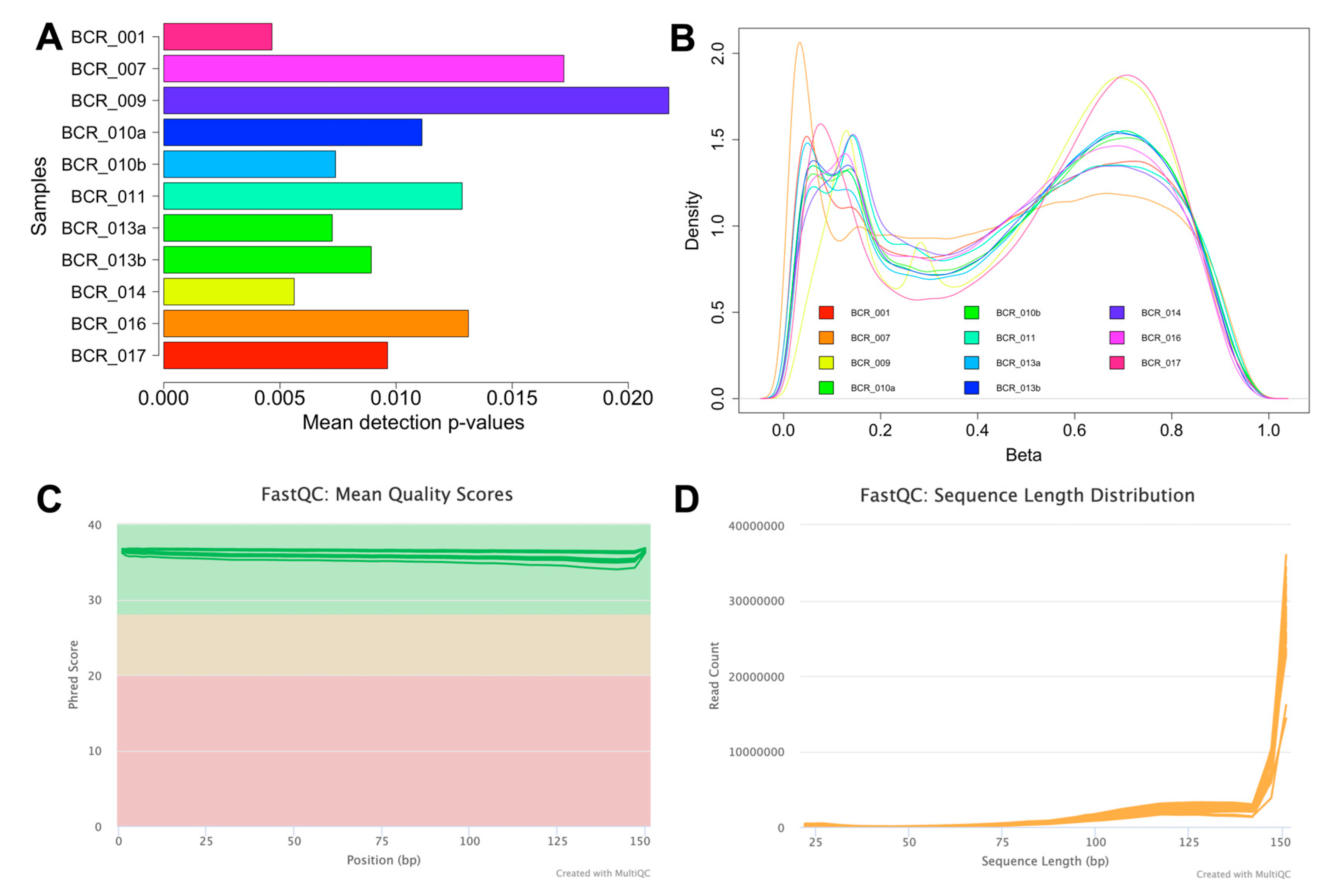
To ensure the accuracy and reliability of the array data, thorough quality assessments were performed. The initial signal quality assessment revealed that out of the total 19 methylomes (17 samples plus two replicates), eight samples had a mean detection p-value > 0.05, indicating poor signal quality, likely due to degraded FFPE material. As a result, these low-quality samples were excluded from further analyses. The remaining 11 samples, which demonstrated reliable signal quality with a mean detection p-value < 0.05 (Figure 2A), were selected for subsequent analysis and are presented in Table 2. The number of probes with mean detection p-values < 0.05 and < 0.01 was then assessed for these samples, as summarised in Table 2. The variation in the number of probes between the two detection p-value thresholds ranged from 1.06–3.47% (9932-32,513 probes). To minimise the loss of probes, a p-value threshold of <0.05 was used for the remaining analysis. Following this, the data were normalised to correct for technical biases and filtered to include only probes with a mean detection p-value < 0.05 (Table 2). Probes located on sex chromosomes, those containing SNPs, and cross-reactive probes were excluded, resulting in 783,503 probes. A density plot of the beta values was subsequently generated for each sample, and this analysis revealed a bimodal distribution with two distinct peaks at the unmethylated and fully methylated ends (Figure 2B). The EPIC v2.0 array platform is a new platform, and limited data on tumour samples are available for this platform. However, our methylation distribution analysis is consistent with the benchmarking analysis performed on the same platform [20]. In addition, our distribution analysis showed higher levels of intermediate methylation (25% to 75% methylation) compared to previous methylation studies [21,22]. This is consistent with the design of the EPICv2.0 platform, which includes more probes located in regulatory regions, such as enhancers, that are known to show intermediate methylation values and are consistent with recently published data using this platform [20,23]. Finally, probe-wise differential methylation analysis can be performed to address the underlying biological hypothesis.
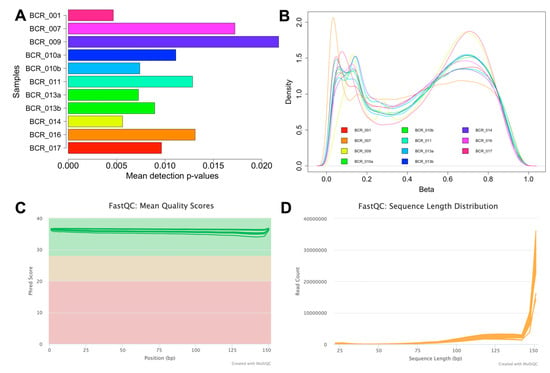
Figure 2.
Quality assessment of the MethylationEPIC v2.0 and RNA-Seq data. (A) A bar plot illustrating the mean detection p-values for the 11 high-quality methylation array samples. Sample IDs are displayed on the Y-axis, and the mean detection p-value of each sample is depicted on the X-axis. (B) Beta value distribution plot showing methylation distribution on a global scale across the 11 high-quality samples. (C) Mean quality scores for RNA-Seq samples after trimming (plotted with Phred scores on the Y-axis vs. read position on the X-axis). The Y-axis is colour-coded into three sections, with green indicating high-quality base calls, yellow for moderate, and red for poor-quality calls. (D) Sequence length distribution plot for RNA samples following trimming, showing the length of sequences in base pairs on the X-axis and the corresponding read counts on the Y-axis.
Table 2.
Summary of probe counts for methylation array data at different mean detection p-value thresholds after filtering.
3.2. RNA-Seq Data Analysis, Assessments and Features
RNA samples (which included one technical replicate) showed a mean DV200 value of 35.13% (see Table 3), which is excellent for FFPE samples. RNA sequencing generated a total of 1.12 billion reads (an average of 62.30 million reads per sample, Table 3). The RNA-Seq data were subjected to further quality assessment checks. The FASTQ reads were trimmed to eliminate adapter sequences and retain high-quality reads with a Phred score > 30 (Figure 2C). Additionally, the sequence length distribution plot indicates high read counts for reads > 140 bp (Figure 2D). The number of reads uniquely mapped to the human genome per sample ranged from 2.33 million to 34.82 million. While the percentage of uniquely mapped reads is low in some samples, the absolute number of mapped reads is sufficient to support downstream analyses, including differential expression and gene biotype assessments. We were also able to quantify the expression of a large number of protein-coding RNAs (7267 to 15,557 genes) and long non-coding RNAs (lncRNAs). Further, we also identified a small proportion of other non-coding RNAs (ncRNAs) that include non-coding RNAs not classified as lncRNA in these clinical samples (Table 3).
Table 3.
Overview of RNA sequencing data analysis, alignment, and features for each sample.
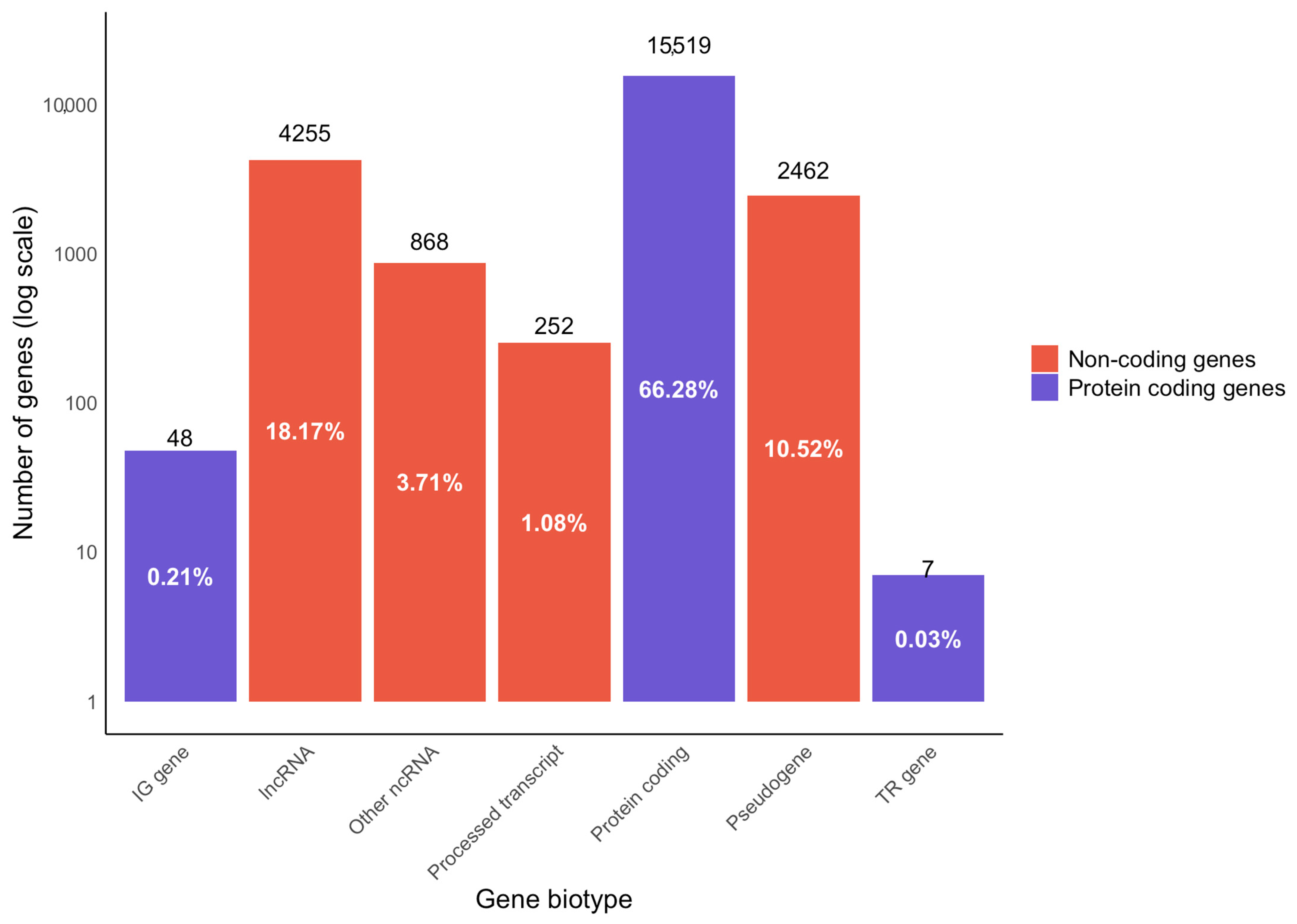
During library preparation, ribosomal RNA (rRNA) was depleted using the Ribo-Zero Gold kit. Our analysis showed < 6% rRNA mapping of the samples (except for BCR_005, which had 10.61% rRNA mapping; Table 3), demonstrating effective rRNA depletion. We have also assessed the features of RNA-Seq data and analysed different biotypes using the Ensembl biotype classification for genes and transcripts (Figure 3). Among the genes observed and analysed in the dataset, 66.28% of the analysed genes were protein coding, which included 0.21% immunoglobulin genes, and 0.03% were genes for T-cell receptors. The remaining non-protein-coding genes encompassed 18.17% lncRNA, 10.52% pseudogenes and 3.71% other ncRNA (comprising microRNA (miRNA), small nucleolar RNA (snoRNA), miscellaneous RNA (misc_RNA), ribosomal RNA (rRNA), small nuclear RNA (snRNA), mitochondrial rRNA (mt_rRNA) and mitochondrial transfer RNA (mt_tRNA)), and 1.08% processed transcripts. Further, it is known that a variety of bacteria are known to influence carcinogenesis [24], and several previous analyses of FFPE RNA-Seq data demonstrated the presence of bacteria in cancer samples, including large datasets in The Cancer Genome Atlas (TCGA) [25,26]. Therefore, we have assessed the overrepresented sequences in our RNA-Seq data and found Enterobacter, Phyllobacterium and Mesorhizobium to be among the top three bacterial genera present. The detected microbiome sequences are reported as observations but are not interpreted as having a causal role in carcinogenesis. For the quantification of expression, only uniquely aligned sequences to the human genome were used. In conjunction with the technical replicates, our FFPE RNA-Seq data produced robust transcriptome quantification.
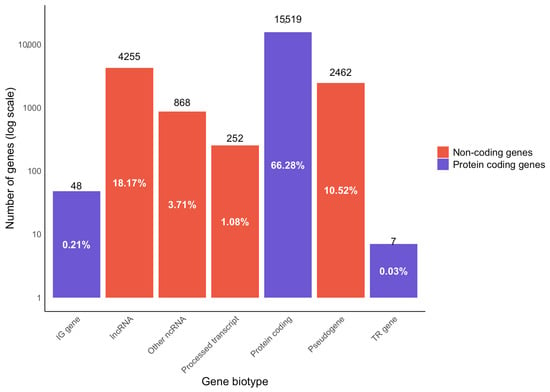
Figure 3.
Gene biotype analysis from RNA-Seq data of PCa FFPE samples. Data showing the distribution of the percentage of genes mapped to protein-coding and non-coding regions of the genome. The protein-coding genes include those that code for immunoglobulins (IG gene) and T-cell receptors (TR gene). The protein non-coding genes encompass pseudogenes, long non-coding RNA (lncRNA), processed transcripts, and other non-coding RNA (ncRNA). The ‘other ncRNAs’ category encompassed consisting of microRNA (miRNA), small nucleolar RNA (snoRNA), miscellaneous RNA (misc_RNA), ribosomal RNA (rRNA), small nuclear RNA (snRNA), mitochondrial rRNA (mt_rRNA) and mitochondrial transfer RNA (mt_tRNA).
3.3. Technical Reproducibility of DNA Methylome and Transcriptome Data
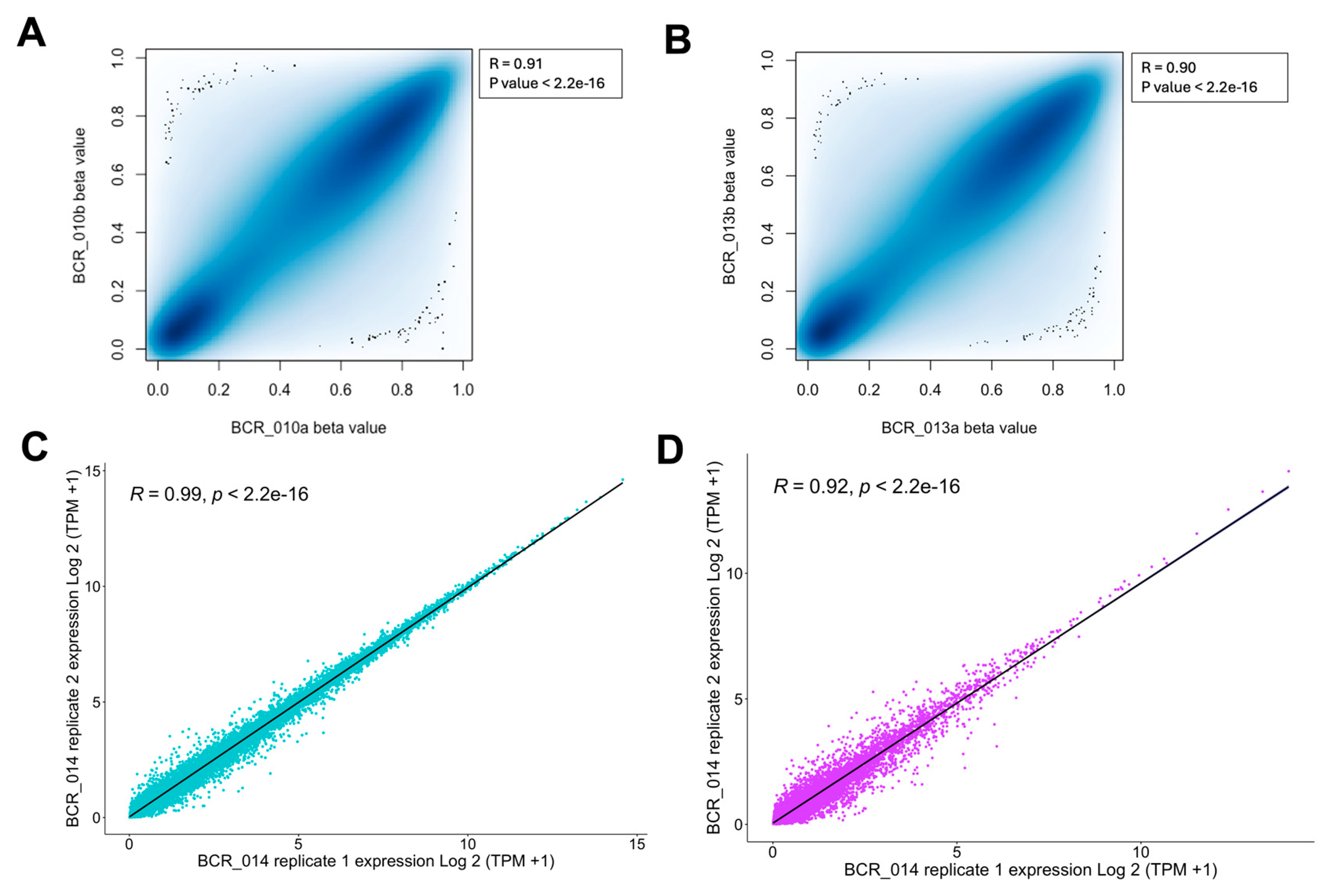
To evaluate the technical reproducibility of the MethylationEPIC v2.0 array data, we have included technical replicates for two samples (BCR_010 and BCR_013) in our study. Consistent experimental conditions were maintained when generating the array data for these replicates. Beta values, representing DNA methylation levels, were calculated for each sample, and their distribution was assessed using the Shapiro-Wilk test. A p-value < 0.05 from the Shapiro-Wilk test indicated that the beta values did not follow a normal distribution, necessitating non-parametric analysis. Therefore, Spearman’s correlation analysis was applied to assess the concordance between the replicates. This analysis revealed a significant correlation in global DNA methylation levels between the replicates for BCR_010 (Spearman’s ρ = 0.91, p-value < 2.2 × 10−16; Figure 4A) and BCR_013 (Spearman’s ρ = 0.90, p-value < 2.2 × 10−16; Figure 4B), indicating high reproducibility between the clinical FFPE samples.
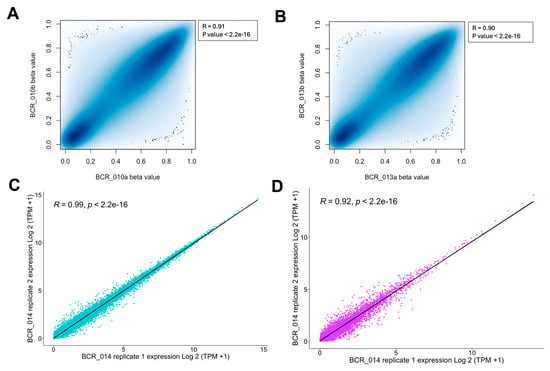
Figure 4.
Analysis of technical reproducibility and features of EPIC v2.0 Array and RNA-Seq platforms for FFPE tissues in prostate cancer. (A) p-value detection distribution for methylation analysis. (B) The beta values of each sample were assessed for normality using the Shapiro-Wilk test, and Spearman’s correlation was conducted to illustrate the technical reproducibility of DNA methylation levels. (C,D) Technical replicate plots for coding and non-coding RNA and correlation measurements.
For RNA-Seq data, we included one technical replicate (BCR_014) and analysed the reproducibility of coding and non-coding RNA separately. We found that the replicates for BCR_014 showed excellent correlation for both coding (Spearman’s ρ = 0.99, p-value < 2.2 × 10−16; Figure 4C) and non-coding RNA (Spearman’s ρ = 0.92, p-value < 2.2 × 10−16; Figure 4D). To our knowledge, our work is the first to assess the technical reproducibility of FFPE samples in coding and non-coding regions from clinical samples.
4. Methods
4.1. Patient Recruitment and Tumour Sample Collection
Participants were recruited from the clinical records of men who underwent radical prostatectomy at Dunedin Hospital (Dunedin, New Zealand) between April 2018 and February 2020. Patients over 18 years of age were eligible for inclusion if they had a diagnosis of histologically confirmed PCa for which they had undergone curative intent radical prostatectomy without routine adjuvant treatment, with a minimum follow-up period of two years post-surgery, available PSA levels for review, and the ability to provide informed consent. A total of 23 eligible participants were identified for potential inclusion, 18 of whom provided written informed consent. A single participant was excluded as no stored tissue sample was available for retrieval. Formalin-fixed paraffin-embedded (FFPE) PCa samples resected via radical prostatectomy (n = 17) were obtained from Awanui Laboratories (Dunedin, New Zealand). Relevant demographic, clinical, and histopathological data were retrospectively obtained from medical records and pathology reports. Follow-up data were available for all participants to at least four years post-surgery. Recruited patients were stratified into respective “Biochemical Recurrence” and “No Recurrence” groups based on the presence or absence of BCR at four years post-operatively, defined as a PSA rise to ≥0.20 ng/mL following prostatectomy (Table 4). A single participant (BCR_001) experienced “Biochemical Persistence” post-operatively, wherein their first post-operative PSA level was raised to ≥0.20 ng/mL. In Table 4, this participant has been excluded from both the recurrent and non-recurrent groups, as they belong instead to a category of their own. All other participants reached a post-operative PSA nadir of <0.05 ng/mL.
Table 4.
Summary of demographics and clinical data for biochemical recurrence, biochemical persistence, and no recurrence cohorts.
4.2. Clinical Data Collection and Description of Clinical Features
Summarised and detailed analysis of clinical data is presented in Table 4. The baseline demographics were similar across all groups. Participants with BCR were younger, with a lower median age (63 versus 69), compared to those without recurrence. All participants were of European ancestry and 7 patients had a documented family history of prostate cancer. The median PSA level at diagnosis was higher for those who developed BCR—8.4 ng/mL (interquartile range (IQR) 5.7–11.2 ng/mL) versus 5.7 ng/mL (IQR 3.0–8.4 ng/mL). PSA density, calculated as the PSA level at diagnosis divided by estimated prostate size on biopsy, was also higher in the BCR group—0.18 ng/mL2 (IQR 0.12–0.25 ng/mL2) versus 0.15 ng/mL2 (IQR 0.12–0.18 ng/mL2). The rates of abnormal digital rectal examinations were similar across the groups.
At the time of transrectal ultrasound-guided (TRUS) biopsy, the majority of tumours detected across all groups were intermediate-risk International Society of Urological Pathology (ISUP) Grade Group 2 or 3 (Gleason 7) in nature, with no high-risk ISUP 5 disease noted [27]. The rates of bilateral tumour involvement (87.5% versus 62.5%), percentage of cores involved (43% versus 27%), and overall percentage of tumour tissue (15% versus 6.3%) were higher in the BCR group. Histological analysis of radical prostatectomy samples confirmed a similar proportion of largely ISUP 2 and 3 tumours across both groups. Both groups had a single patient upstaged to ISUP 5 disease. Notably, none of the patients in the BCR group had low-risk ISUP 1 disease on final histology. The rates of seminal vesicle invasion, lymphovascular invasion, and extracapsular extension were low in both groups. One participant in the BCR group had a positive surgical margin. There were no significant differences between the groups with respect to the time from diagnosis to radical prostatectomy.
The median time to BCR was 2.2 years (IQR 1.8–2.6 years). PSA doubling time (PSA-DT), the estimated time for PSA to increase two-fold, was calculated using the nomogram created by Pound et al. [28]. The median PSA-DT post-prostatectomy for patients experiencing recurrence was 5.8 months (IQR 5.1–6.5 months). Of this cohort of BCR patients, 50% demonstrated imaging-confirmed disease progression at the time of four-year follow-up in the form of either local recurrence (37.5%), regional nodal metastasis (37.5%), or distant metastasis (25%). All these patients received salvage radiation therapy at the point of confirmed recurrence, and 75% had further recurrence warranting additional radiation therapy, with or without adjuvant androgen-deprivation therapy. Despite no imaging confirmation of disease recurrence, an additional patient (BCR_013) from the BCR cohort also underwent salvage radiotherapy for presumed local recurrence after experiencing early BCR within 10 months of surgery. The participant with biochemical persistence also received salvage radiation therapy and subsequent androgen deprivation for further recurrence.
Several recorded variables have been suggested to hold prognostic value for tumour progression and recurrence. Surgical margin status and PSA-DT possess predictive power as standalone variables [29,30]. Pre-operative PSA, surgical margin status, extracapsular extension, seminal vesicle invasion, and lymph node invasion on radical prostatectomy have reported improved predictive power when considered alongside one another [31,32].
4.3. DNA Extraction and Genome-Wide Methylation Profiling
DNA was extracted from FFPE tissues (n = 17, including technical replicates of two samples). A 10 μm section from the top of the FFPE was discarded (as this section was exposed to air). Following this, eight 10 μm sections were used for DNA extraction [33]. The extraction was performed using a modified protocol of the QIAamp DNA FFPE Kit (Qiagen, Hilden, Germany) in conjunction with a supplementary deparaffinisation solution protocol. DNA was quantified using the Qubit 1X dsDNA BR kit and Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). The samples were then sent to the Australian Genome Research Facility (Melbourne, VIC, Australia) for methylation array data generation. A second round of quantification and quality checks was performed by the facility using the QuantiFluor dsDNA System (Promega, Fitchburg, WI, USA) and 1% E-gel, respectively. Approximately 250 ng of genomic DNA was bisulfite-converted using the Zymo EZ-96 DNA Methylation kit (Zymo Research, Irvine, CA, USA) and restored using the Illumina Infinium HD FFPE DNA Restoration kit (Illumina, CA, USA). Samples were then processed on Illumina Human Methylation EPIC version 2.0 BeadChips (Illumina, CA, USA).
4.4. DNA Methylome (MethylationEPIC v2.0) Data Analysis
The raw data from the Illumina Human Methylation EPIC v2.0 array was obtained in the Illumina Data format (IDAT). For each sample, the raw data consisted of two files: grn.idat (green channel intensity) and red.idat (red channel intensity), which captured the intensity levels necessary for methylation analysis. The IDAT files were processed using the minfi package as part of a cross-package workflow in RStudio [34,35]. For EPIC v2.0, we have used the IlluminaHumanMethylationEPICv2anno.20a1.hg38 package for probe annotations.
The minfi package was then used to convert the raw red and green channel intensities into methylated (M) and unmethylated (U) signal intensities [36]. This conversion is a key step in the preprocessing pipeline, as it provides the foundational data needed to calculate methylation levels across the genome. Next, the RnBeads package was utilised to check the quality of the data [37]. The detection p-value was calculated for each sample by comparing the total signal intensity (M + U) for each probe to the background signal level, which was estimated from the negative control probes designed to measure non-specific binding. Samples with reliable signals (p-value < 0.05) were further filtered to exclude probes overlapping with single nucleotide polymorphisms (SNPs) identified by Pidsley et al. (2016) [38]. Any samples that had a detection p-value > 0.05 were not included in further analyses (see Table 2).
The functional normalisation method from the minfi package was used to minimise technical variation [39,40,41]. This was followed by retaining only those probes with a mean detection p-value < 0.05, across all samples. Probes with SNPs at the CpG interrogation site or single nucleotide extension were removed using the dropLociWithSnps function in minfi [36], along with probes showing cross-hybridisation [38]. Beta values were calculated for each CpG probe that was retained after the filtering steps for each sample using the formula [M/(U + M + 100)]. The addition of 100 to the denominator served as a small background correction, yielding a beta value ranging from 0 to 1 (corresponding to 0% to 100% methylation, respectively, at each CpG site). Spearman’s correlation tests were performed on the filtered probes using beta values to assess the relationships between replicates.
4.5. RNA Extraction and RNA Sequencing
RNA was extracted from the same FFPE tissue blocks (n = 17, including a technical replicate for each sample) used for DNA extraction. The first 10 μm section exposed to air was discarded, and the next eight 10 μm sections were used for extraction. Total RNA was extracted immediately using a modified Qiagen RNeasy FFPE Kit protocol (Qiagen, Hilden, Germany). RNA was quantified using a Qubit RNA BR kit (Thermo Fisher Scientific, MA, USA). TruSeq stranded total RNA libraries were prepared by AgResearch Invermay (Mosgiel, New Zealand) using the TruSeq RNA Library Prep Kit v2 (Illumina, CA, USA), and rRNA was depleted using the Ribo-Zero Gold kit (Illumina, CA, USA) according to the manufacturer’s protocol. The quality of the libraries was checked on an Illumina iSeq flow cell (Illumina, CA, USA), and sequencing was performed on two flow cell lanes of an Illumina NovaSeq (Illumina, CA, USA) generating 150 bp paired-end reads.
4.6. RNA-Sequencing and Data Analysis
RNA-Seq reads were evaluated for quality using FastQC v0.12.1, and summarised with the MultiQC v1.12 tool [42]. Trim Galore v0.6.10 was used to remove adapter sequences and filter out low-quality reads. Trimming was performed with a Phred quality score threshold of 30 and default settings, which included adapter auto-detection. The FastQ Screen v0.15.3 [43] tool was utilised to analyse overrepresented sequences and identify potential contamination in the trimmed reads by aligning the reads to the default databases of the FastQ Screen tool (downloaded using fastq_screen --get_genomes function). The trimmed reads were then aligned to the following human rRNA reference sequences: NR_023363, NR_003285, NR_003286 and NR_003287, using Bowtie2 v2.5.1 aligner [44] to determine the proportion of reads mapped to rRNA, allowing for the assessment of rRNA depletion. The reads were subsequently mapped to the human genome using STAR aligner v2.7.11b [45]. While performing STAR alignment, the Ensembl Homo sapiens GRCh37 reference genome (with version 87 annotation) was used with default settings. STAR alignment generated SAM files, which were converted into BAM files and sorted using Samtools v1.19.2 [46]. Gene counts for each gene ID were obtained using featureCounts v2.0.6, with BAM files as the input [47]. The featureCounts analysis was conducted using the following parameters: -s 0 for unstranded read counting, -p for paired-end reads, and -g gene_id to count reads mapped to gene IDs, using the Ensembl Homo sapiens GRCh37 version 87 annotation file. Subsequently, genes with very low counts were excluded by retaining only genes with Counts Per Million (CPM) > 0.5 in at least nine samples (i.e., 50% of the samples), and then count values were normalised to Transcripts Per Million (TPM). The R package biomaRt v2.54.1 [48,49] was used to retrieve gene names and gene biotypes using the hsapiens_gene_ensembl database (accessed via the Ensembl GRCh37, URL: grch37.ensembl.org). Common protein-coding and non-coding genes were identified between replicates, and correlation analysis was independently performed for common protein-coding and non-coding genes. Spearman correlation analyses were conducted separately for common protein-coding and non-coding genes in the replicates using log2(TPM + 1) values for each gene biotype. A step-by-step description of RNA-Seq analysis from clinical samples has been published previously [50,51,52,53], and we have followed these steps for processing these data.
5. User Notes
Molecular biomarkers have shown potential as prospective tools for cancer diagnosis and monitoring disease progression. Here, we provide genome-scale DNA methylome and whole transcriptome data samples for patients with or without evidence of BCR after prostatectomy using cutting-edge assays. The RNA-Seq and array-based DNA methylome data are available to use from the GEO database GSE282574. All bioinformatics analyses can be conducted on the most common operating systems (Windows, Linux, and Mac).
Recent technological advances have facilitated a growing focus on the use of molecular biomarkers as novel tools for cancer diagnosis and disease monitoring. For example, genetic changes have been implicated in numerous cancer types and form the basis of many novel screening and diagnostic tools. In PCa, however, a paucity of identifiable genetic drivers of disease has meant that genetic mutations have shown modest clinical utility thus far [54]. However, other molecular changes, such as alterations in gene expression and DNA methylation profiles, have been heavily implicated in cancer, including PCa progression, and could be further exploited to identify biomarkers for PCa risk stratification [55,56]. Identification of such biomarkers associated with disease progression would have the scope to identify high-risk individuals at earlier disease stages, allowing for more appropriate treatment selection and post-treatment monitoring to improve clinical outcomes. Importantly, we provide detailed data for each sample to make clinical correlations with multi-omic data. This is a major strength of this work, as the majority of the previous multi-omic work focused on generating data with limited clinical information, which prohibits downstream clinical utility. Further, global and targeted epigenomic editing methods are emerging rapidly [57,58,59,60], and identifying novel candidates provides future opportunities to explore their role in BCR biology and for developing potential new therapeutic targets.
The majority of the clinical tumour samples are stored as FFPE, and FFPE material poses a challenge for generating genome-scale data [61]. For our methylome analysis, we have identified samples that showed lower signal intensity. This suggests that although array-based methylome analysis provides a restoration method for obtaining data from FFPE samples, it may not be sufficient to obtain good-quality data for every sample. Therefore, further development of methylation analysis using FFPE samples is required. For RNA sequencing, we were able to obtain good quality and reproducible data as described. However, for some samples, the mapping levels were low. The challenges of using FFPE samples for RNA-Seq data have also been described in previous prostate cancer studies [62,63]. Improvement in the analysis of FFPE samples will, therefore, remain a subject of research for better clinical translation. It is important to note that our study is based on a relatively small sample size (n = 17). This may limit the generalisability of our findings and the statistical power of our analyses. Future prospective studies with larger sample sizes would be valuable to validate and extend the discovery of epigenetic and transcriptomic markers of prostate cancer. A larger cohort would further enhance the robustness of the identified biomarkers and the development of effective risk stratification tools with potential clinical utility. Our data contribute to the growing amount of omics data aimed at predicting outcomes and patient benefits [64].
Epigenetic and expression markers of BCR can aid in developing a biomarker panel capable of predicting the risk of BCR at the time of diagnosis or radical prostatectomy. The application of these biomarker panels would help inform clinical decision-making as adjunct risk stratification tools and provide clinical benefits through more accurate treatment selection and monitoring. Our work will contribute to the basis of developing a molecular test that can accurately predict the risk of PCa recurrence by analysing molecular changes already present at the time of surgery. We hope that such a test can be used to improve risk stratification for PCa patients and improve health outcomes through a more appropriate selection of clinical treatment options. This work will complement ongoing research in this space and contribute to building a ‘complete picture’ of the molecular landscape and an eventual drive towards providing individualised and equitable PCa care for patients.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/data9120150/s1, Table S1: Patient clinical data.
Author Contributions
Conceptualisation, J.S., A.D.Z., E.J.R. and A.C.; methodology, formal analysis, J.S., A.Y., E.J.R., P.A., S.S.V., A.D., G.G. and A.C.; investigation, J.S., A.Y., E.J.R., P.A., S.S.V., A.D., G.G. and A.C.; resources, E.J.R., A.D.Z., R.M.S. and A.C.; data curation, J.S., A.Y., E.J.R., P.A., S.S.V., A.D. and A.C.; writing—original draft preparation, J.S., A.Y. and A.D.; writing—review and editing, All authors; visualisation, All authors; supervision, E.J.R., J.S. and A.C.; project administration, E.J.W., E.J.R., J.S. and A.C.; funding acquisition, A.C., E.J.R., J.S., E.J.W., R.M.S. and A.D.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Healthcare Otago Charitable Trust (J.S. and E.J.W.) and TD Scott Chair in Urology (A.D.Z. and A.C.).
Institutional Review Board Statement
This study was approved by the Southern Health and Disability Ethics Committee (ethics number: 21/STH/184).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data have been deposited in the GEO database under accession number GSE282574.
Acknowledgments
The authors are firstly grateful to the patients and their whānau who have contributed to this work. We thank Anamika Ghosh, Lesley Marriott, Alastair Hepburn, John Woodfield, and the Department of Urology at Dunedin Hospital (Health New Zealand Te Whatu Ora—Southern) for their role in patient recruitment. We also acknowledge Awanui Laboratories and the Histology Unit (Department of Pathology, University of Otago) for their assistance with sample retrieval and processing.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Kupelian, P.A.; Buchsbaum, J.C.; Elshaikh, M.; Reddy, C.A.; Zippe, C.; Klein, E.A. Factors affecting recurrence rates after prostatectomy or radiotherapy in localized prostate carcinoma patients with biopsy Gleason score 8 or above. Cancer 2002, 95, 2302–2307. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.E.; Yousefi, K.; Davicioni, E.; Ghadessi, M.; Johnson, M.H.; Sundi, D.; Tosoian, J.J.; Han, M.; Humphreys, E.B.; Partin, A.W.; et al. Utility of Risk Models in Decision Making After Radical Prostatectomy: Lessons from a Natural History Cohort of Intermediate- and High-Risk Men. Eur. Urol. 2016, 69, 496–504. [Google Scholar] [CrossRef]
- Cookson, M.S.; Aus, G.; Burnett, A.L.; Canby-Hagino, E.D.; D’Amico, A.V.; Dmochowski, R.R.; Eton, D.T.; Forman, J.D.; Goldenberg, S.L.; Hernandez, J. Variation in the definition of biochemical recurrence in patients treated for localized prostate cancer: The American Urological Association Prostate Guidelines for Localized Prostate Cancer Update Panel report and recommendations for a standard in the reporting of surgical outcomes. J. Urol. 2007, 177, 540–545. [Google Scholar]
- Heidenreich, A.; Bastian, P.J.; Bellmunt, J.; Bolla, M.; Joniau, S.; van der Kwast, T.; Mason, M.; Matveev, V.; Wiegel, T.; Zattoni, F. EAU guidelines on prostate cancer. Part II: Treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur. Urol. 2014, 65, 467–479. [Google Scholar] [CrossRef]
- Shore, N.D.; Moul, J.W.; Pienta, K.J.; Czernin, J.; King, M.T.; Freedland, S.J. Biochemical recurrence in patients with prostate cancer after primary definitive therapy: Treatment based on risk stratification. Prostate Cancer Prostatic Dis. 2024, 27, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Van den Broeck, T.; Van Den Bergh, R.C.; Arfi, N.; Gross, T.; Moris, L.; Briers, E.; Cumberbatch, M.; De Santis, M.; Tilki, D.; Fanti, S. Prognostic value of biochemical recurrence following treatment with curative intent for prostate cancer: A systematic review. Eur. Urol. 2019, 75, 967–987. [Google Scholar] [CrossRef]
- Preisser, F.; Abrams-Pompe, R.S.; Stelwagen, P.J.; Böhmer, D.; Zattoni, F.; Magli, A.; Rivas, J.G.; Dilme, R.V.; Sepulcri, M.; Eguibar, A. European association of urology biochemical recurrence risk classification as a decision tool for salvage radiotherapy—A multicenter study. Eur. Urol. 2024, 85, 164–170. [Google Scholar] [CrossRef]
- Falagario, U.G.; Abbadi, A.; Remmers, S.; Björnebo, L.; Bogdanovic, D.; Martini, A.; Valdman, A.; Carrieri, G.; Menon, M.; Akre, O. Biochemical recurrence and risk of mortality following radiotherapy or radical prostatectomy. JAMA Netw. Open 2023, 6, e2332900. [Google Scholar] [CrossRef]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.-J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Ezegbogu, M.; Wilkinson, E.; Reid, G.; Rodger, E.J.; Brockway, B.; Russell-Camp, T.; Kumar, R.; Chatterjee, A. Cell-free DNA methylation in the clinical management of lung cancer. Trends Mol. Med. 2024, 30, 499–515. [Google Scholar] [CrossRef]
- Smith, J.; Barnett, E.; Rodger, E.J.; Chatterjee, A.; Subramaniam, R.M. Neuroendocrine Neoplasms: Genetics and Epigenetics. PET Clin. 2023, 18, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Rodger, E.J.; Gimenez, G.; Ajithkumar, P.; Stockwell, P.A.; Almomani, S.; Bowden, S.A.; Leichter, A.L.; Ahn, A.; Pattison, S.; McCall, J.L.; et al. An epigenetic signature of advanced colorectal cancer metastasis. iScience 2023, 26, 106986. [Google Scholar] [CrossRef]
- Banerjee, R.; Smith, J.; Eccles, M.R.; Weeks, R.J.; Chatterjee, A. Epigenetic basis and targeting of cancer metastasis. Trends Cancer 2022, 8, 226–241. [Google Scholar] [CrossRef]
- FitzGerald, L.M.; Jung, C.-h.; Wong, E.M.; Joo, J.E.; Bassett, J.K.; Dowty, J.G.; Wang, X.; Dai, J.Y.; Stanford, J.L.; O’Callaghan, N.; et al. Detection of differentially methylated CpGs between tumour and adjacent benign cells in diagnostic prostate cancer samples. Sci. Rep. 2024, 14, 17877. [Google Scholar] [CrossRef] [PubMed]
- Savio, A.J.; Kamdar, S.; Jeyapala, R.; Olkhov-Mitsel, E.; Cuizon, C.; Finelli, A.; Zlotta, A.R.; Toi, A.; Fleshner, N.E.; van der Kwast, T.; et al. Methylation Markers in Prostate Biopsies Are Prognosticators for Late Biochemical Recurrence and Therapy after Surgery in Prostate Cancer Patients. J. Mol. Diagn. 2020, 22, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.G.; Chen, W.S.; Li, H.; Foye, A.; Zhang, M.; Sjöström, M.; Aggarwal, R.; Playdle, D.; Liao, A.; Alumkal, J.J. The DNA methylation landscape of advanced prostate cancer. Nat. Genet. 2020, 52, 778–789. [Google Scholar] [CrossRef]
- Ramesh, A.; Chatterjee, A.; Subramaniam, R.M. Neuroendocrine Neoplasms: Epidemiology, Diagnosis, and Management. PET Clin. 2023, 18, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Barnett, E.; Smith, J.; Wilkinson, E.; Subramaniam, R.M.; Zarrabi, A.; Rodger, E.J.; Chatterjee, A. Genetic and epigenetic features of neuroendocrine prostate cancer and their emerging applications. Int. Rev. Cell Mol. Biol. 2024, 383, 41–66. [Google Scholar] [CrossRef]
- Peters, T.J.; Meyer, B.; Ryan, L.; Achinger-Kawecka, J.; Song, J.; Campbell, E.M.; Qu, W.; Nair, S.; Loi-Luu, P.; Stricker, P.; et al. Characterisation and reproducibility of the HumanMethylationEPIC v2.0 BeadChip for DNA methylation profiling. BMC Genom. 2024, 25, 251. [Google Scholar] [CrossRef]
- Chatterjee, A.; Stockwell, P.A.; Rodger, E.J.; Morison, I.M. Genome-scale DNA methylome and transcriptome profiling of human neutrophils. Sci. Data 2016, 3, 160019. [Google Scholar] [CrossRef]
- Chatterjee, A.; Stockwell, P.A.; Rodger, E.J.; Duncan, E.J.; Parry, M.F.; Weeks, R.J.; Morison, I.M. Genome-wide DNA methylation map of human neutrophils reveals widespread inter-individual epigenetic variation. Sci. Rep. 2015, 5, 17328. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Scholer, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.R.; Bleich, R.M.; Arthur, J.C. Microbiota Effects on Carcinogenesis: Initiation, Promotion, and Progression. Annu. Rev. Med. 2021, 72, 243–261. [Google Scholar] [CrossRef]
- Strong, M.J.; Xu, G.; Morici, L.; Splinter Bon-Durant, S.; Baddoo, M.; Lin, Z.; Fewell, C.; Taylor, C.M.; Flemington, E.K. Microbial contamination in next generation sequencing: Implications for sequence-based analysis of clinical samples. PLoS Pathog. 2014, 10, e1004437. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; Crabtree, J.; Mattick, J.S.; Anderson, K.E.; Dunning Hotopp, J.C. Distinguishing potential bacteria-tumor associations from contamination in a secondary data analysis of public cancer genome sequence data. Microbiome 2017, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Egevad, L.; Amin, M.B.; Delahunt, B.; Srigley, J.R.; Humphrey, P.A.; Grading, C. The 2014 International Society of Urological Pathology (ISUP) consensus conference on Gleason grading of prostatic carcinoma: Definition of grading patterns and proposal for a new grading system. Am. J. Surg. Pathol. 2016, 40, 244–252. [Google Scholar] [CrossRef]
- Pound, C.R.; Partin, A.W.; Eisenberger, M.A.; Chan, D.W.; Pearson, J.D.; Walsh, P.C. Natural history of progression after PSA elevation following radical prostatectomy. JAMA 1999, 281, 1591–1597. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.G.; Blute, M.L.; Bergstralh, E.J.; Slezak, J.M.; Zincke, H. PSA doubling time as a predictor of clinical progression after biochemical failure following radical prostatectomy for prostate cancer. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2001; pp. 576–581. [Google Scholar]
- Budäus, L.; Isbarn, H.; Eichelberg, C.; Lughezzani, G.; Sun, M.; Perrotte, P.; Chun, F.K.H.; Salomon, G.; Steuber, T.; Köllermann, J. Biochemical recurrence after radical prostatectomy: Multiplicative interaction between surgical margin status and pathological stage. J. Urol. 2010, 184, 1341–1346. [Google Scholar] [CrossRef]
- Cooperberg, M.R.; Carroll, P.R.; Klotz, L. Active surveillance for prostate cancer: Progress and promise. J. Clin. Oncol. 2011, 29, 3669–3676. [Google Scholar] [CrossRef]
- Cooperberg, M.R.; Cowan, J.E.; Hilton, J.F.; Reese, A.C.; Zaid, H.B.; Porten, S.P.; Shinohara, K.; Meng, M.V.; Greene, K.L.; Carroll, P.R. Outcomes of active surveillance for men with intermediate-risk prostate cancer. J. Clin. Oncol. 2011, 29, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Ludgate, J.L.; Wright, J.; Stockwell, P.A.; Morison, I.M.; Eccles, M.R.; Chatterjee, A. A streamlined method for analysing genome-wide DNA methylation patterns from low amounts of FFPE DNA. BMC Med. Genom. 2017, 10, 54. [Google Scholar] [CrossRef]
- Maksimovic, J.; Phipson, B.; Oshlack, A. A cross-package Bioconductor workflow for analysing methylation array data. F1000Research 2016, 5, 1281. [Google Scholar] [CrossRef]
- Noguera-Castells, A.; García-Prieto, C.A.; Álvarez-Errico, D.; Esteller, M. Validation of the new EPIC DNA methylation microarray (900K EPIC v2) for high-throughput profiling of the human DNA methylome. Epigenetics 2023, 18, 2185742. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef]
- Muller, F.; Scherer, M.; Assenov, Y.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. RnBeads 2.0: Comprehensive analysis of DNA methylation data. Genome Biol. 2019, 20, 55. [Google Scholar] [CrossRef]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef]
- Sala, C.; Di Lena, P.; Fernandes Durso, D.; Prodi, A.; Castellani, G.; Nardini, C. Evaluation of pre-processing on the meta-analysis of DNA methylation data from the Illumina HumanMethylation450 BeadChip platform. PLoS ONE 2020, 15, e0229763. [Google Scholar] [CrossRef] [PubMed]
- Welsh, H.; Batalha, C.; Li, W.; Mpye, K.L.; Souza-Pinto, N.C.; Naslavsky, M.S.; Parra, E.J. A systematic evaluation of normalization methods and probe replicability using infinium EPIC methylation data. Clin. Epigenetics 2023, 15, 41. [Google Scholar] [CrossRef]
- Fortin, J.P.; Labbe, A.; Lemire, M.; Zanke, B.W.; Hudson, T.J.; Fertig, E.J.; Greenwood, C.M.; Hansen, K.D. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014, 15, 503. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Dobin, A.; Gingeras, T.R. Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinform. 2015, 51, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Gimenez, G.; Stockwell, P.A.; Rodger, E.J.; Chatterjee, A. Strategy for RNA-Seq Experimental Design and Data Analysis. Methods Mol. Biol. 2023, 2588, 249–278. [Google Scholar] [CrossRef]
- Chatterjee, A.; Ahn, A.; Rodger, E.J.; Stockwell, P.A.; Eccles, M.R. A Guide for Designing and Analyzing RNA-Seq Data. Methods Mol. Biol. 2018, 1783, 35–80. [Google Scholar] [CrossRef]
- Leichter, A.L.; Purcell, R.V.; Sullivan, M.J.; Eccles, M.R.; Chatterjee, A. Multi-platform microRNA profiling of hepatoblastoma patients using formalin fixed paraffin embedded archival samples. GigaScience 2015, 4, 54. [Google Scholar] [CrossRef]
- Chatterjee, A.; Leichter, A.L.; Fan, V.; Tsai, P.; Purcell, R.V.; Sullivan, M.J.; Eccles, M.R. A cross comparison of technologies for the detection of microRNAs in clinical FFPE samples of hepatoblastoma patients. Sci. Rep. 2015, 5, 10438. [Google Scholar] [CrossRef] [PubMed]
- Sathianathen, N.J.; Kuntz, K.M.; Alarid-Escudero, F.; Lawrentschuk, N.L.; Bolton, D.M.; Murphy, D.G.; Weight, C.J.; Konety, B.R. Incorporating biomarkers into the primary prostate biopsy setting: A cost-effectiveness analysis. J. Urol. 2018, 200, 1215–1220. [Google Scholar] [CrossRef]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Eccles, M.R. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin. Cancer Biol. 2018, 51, 149–159. [Google Scholar] [CrossRef]
- Banerjee, R.; Ajithkumar, P.; Keestra, N.; Smith, J.; Gimenez, G.; Rodger, E.J.; Eccles, M.R.; Antony, J.; Weeks, R.J.; Chatterjee, A. Targeted DNA Methylation Editing Using an All-in-One System Establishes Paradoxical Activation of EBF3. Cancers 2024, 16, 898. [Google Scholar] [CrossRef]
- Smith, J.; Banerjee, R.; Weeks, R.J.; Chatterjee, A. Editing of DNA Methylation Patterns Using CRISPR-Based Tools. Methods Mol. Biol. 2022, 2458, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Urbano, A.; Smith, J.; Weeks, R.J.; Chatterjee, A. Gene-Specific Targeting of DNA Methylation in the Mammalian Genome. Cancers 2019, 11, 1515. [Google Scholar] [CrossRef] [PubMed]
- Nunez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 2021, 184, 2503–2519.e17. [Google Scholar] [CrossRef] [PubMed]
- Arreaza, G.; Qiu, P.; Pang, L.; Albright, A.; Hong, L.Z.; Marton, M.J.; Levitan, D. Pre-Analytical Considerations for Successful Next-Generation Sequencing (NGS): Challenges and Opportunities for Formalin-Fixed and Paraffin-Embedded Tumor Tissue (FFPE) Samples. Int. J. Mol. Sci. 2016, 17, 1579. [Google Scholar] [CrossRef]
- Nikitina, A.S.; Sharova, E.I.; Danilenko, S.A.; Butusova, T.B.; Vasiliev, A.O.; Govorov, A.V.; Prilepskaya, E.A.; Pushkar, D.Y.; Kostryukova, E.S. Novel RNA biomarkers of prostate cancer revealed by RNA-seq analysis of formalin-fixed samples obtained from Russian patients. Oncotarget 2017, 8, 32990–33001. [Google Scholar] [CrossRef] [PubMed]
- Long, Q.; Xu, J.; Osunkoya, A.O.; Sannigrahi, S.; Johnson, B.A.; Zhou, W.; Gillespie, T.; Park, J.Y.; Nam, R.K.; Sugar, L.; et al. Global transcriptome analysis of formalin-fixed prostate cancer specimens identifies biomarkers of disease recurrence. Cancer Res. 2014, 74, 3228–3237. [Google Scholar] [CrossRef]
- Rade, M.; Kreuz, M.; Borkowetz, A.; Sommer, U.; Blumert, C.; Füssel, S.; Bertram, C.; Löffler, D.; Otto, D.J.; Wöller, L.A.; et al. A reliable transcriptomic risk-score applicable to formalin-fixed paraffin-embedded biopsies improves outcome prediction in localized prostate cancer. Mol. Med. 2024, 30, 19. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).