
Sodium–Glucose Cotransporter 2 Inhibitors to Decrease the Uric Acid Concentration—A Novel Mechanism of Action

 ,
,  ,
,  ,
,  and
and 
(This article belongs to the Section Cardiovascular Clinical Research)
Abstract
:1. Introduction
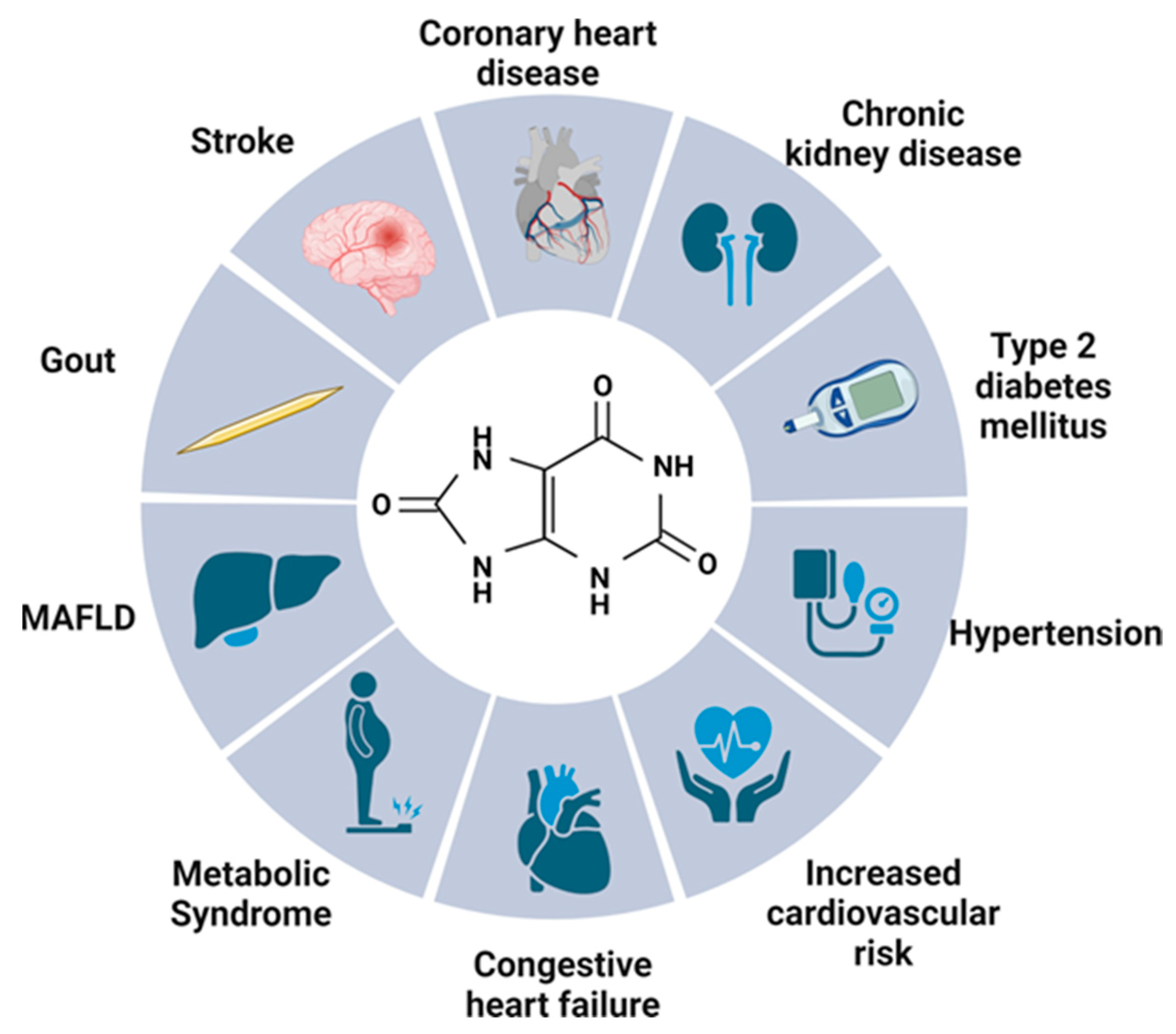
2. Uric Acid—Multifactorial Effects on the Human Body
2.1. Gout
2.2. Kidneys
2.3. Metabolic Syndrome
2.4. Type 2 Diabetes Mellitus
2.5. Neurological Effect
2.6. Cardiovascular Risk
2.6.1. Uric Acid in Cardiovascular Pathology
2.6.2. Coronary Heart Disease, Stroke or Mortality
2.6.3. Congestive Heart Failure
2.6.4. Hypertension
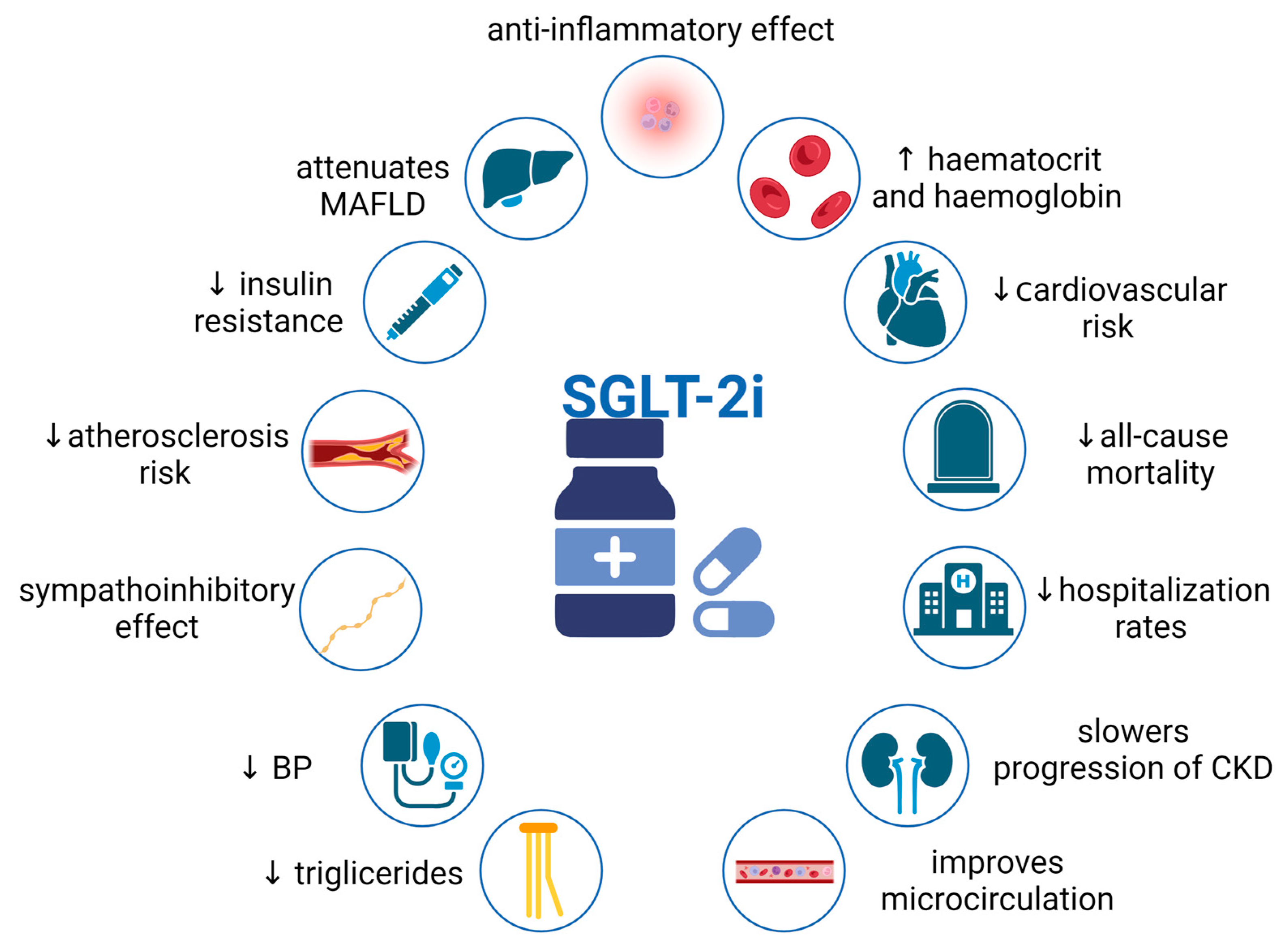
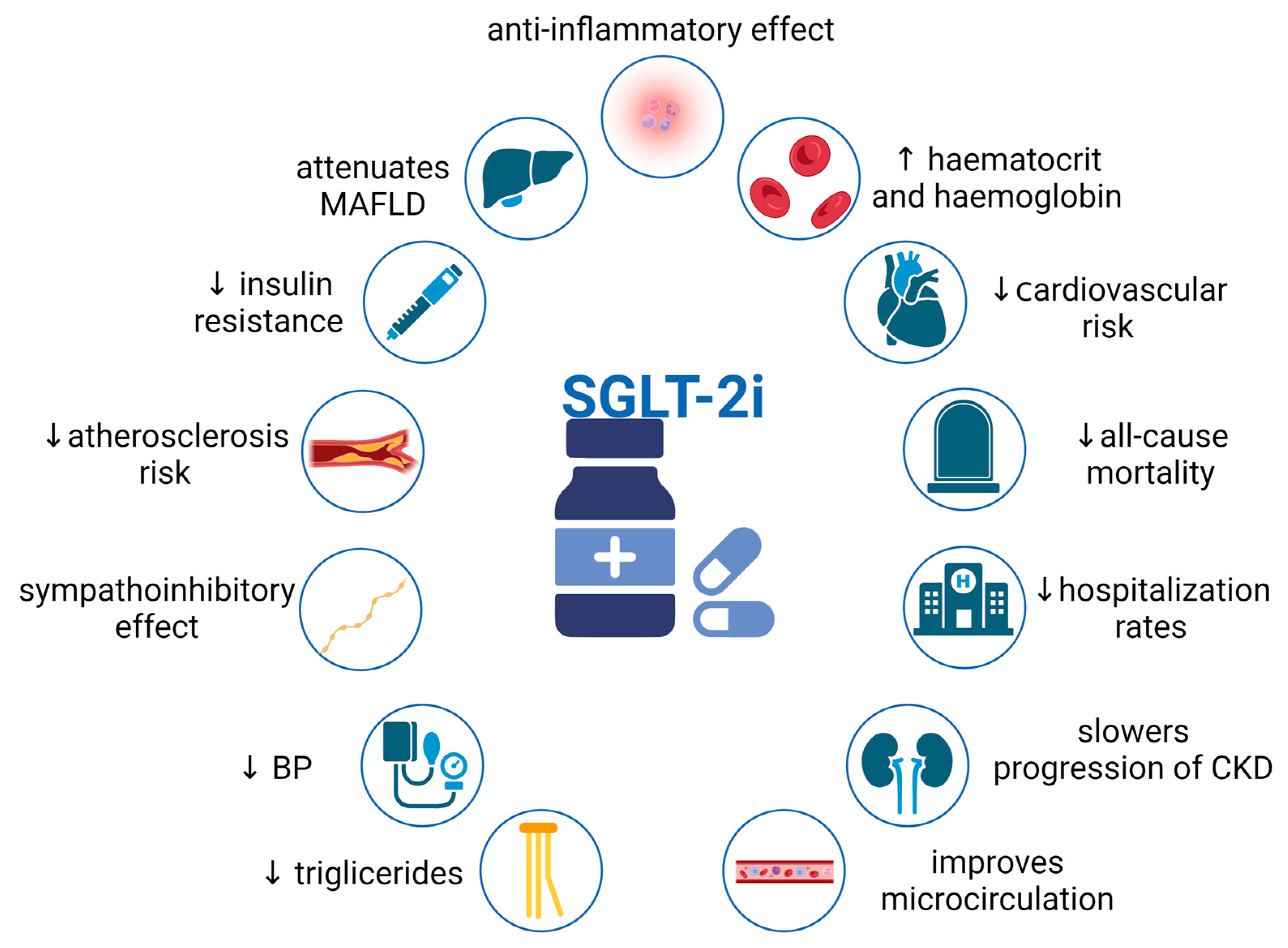
3. Pleiotropic Effects of SGLT2i
3.1. Renal Effects of SGLT2i—Counteraction against Hyperfiltration, Proteinuria, and Renal Fibrosis
3.2. Cardiovascular Effects of SGLT2i
3.2.1. SGLT2i Treatment Decreases Heart Failure Risk
3.2.2. Lipid Profile and Blood Pressure
3.2.3. SGLT2i—Is There Any Influence on the Renin–Angiotensin System?
3.2.4. SGLT2is Change Sympathetic Nervous System Activity
3.2.5. SGLT2is As Myocardium Protectors
3.3. Other Effects
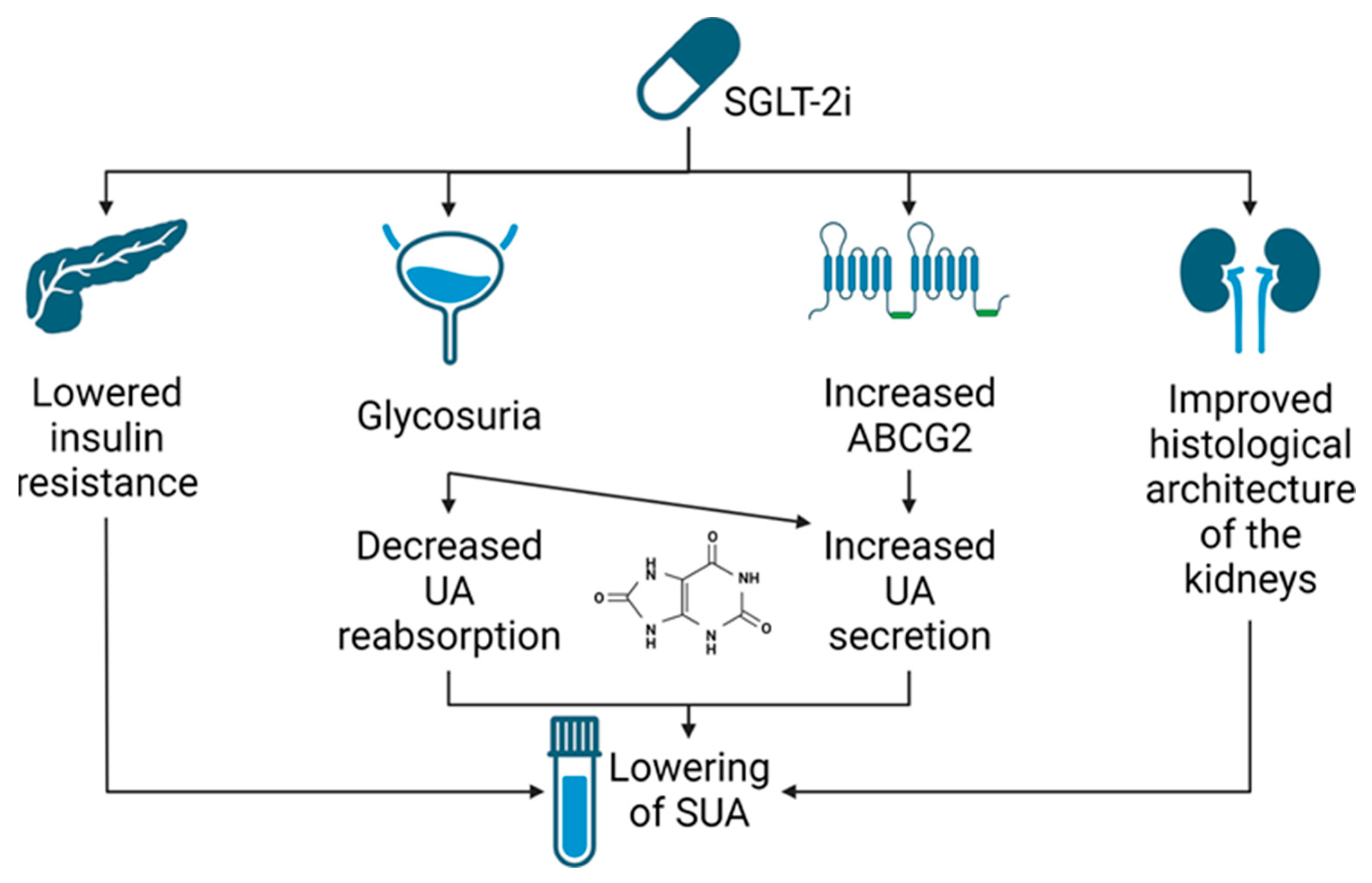
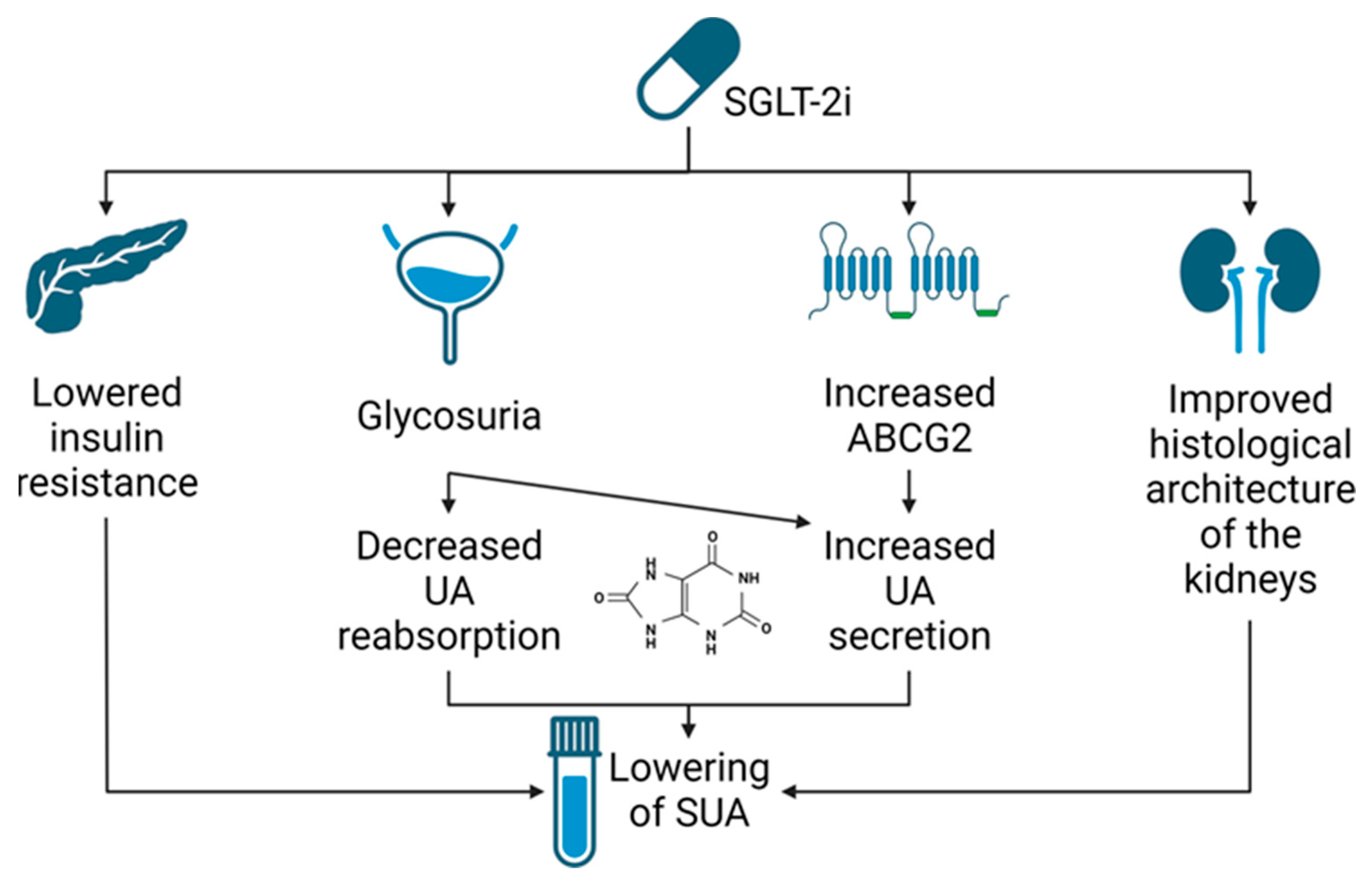
4. SGLT2i—Potential Mechanism of Serum Uric Acid Reduction
4.1. Renal Urate Management
4.2. Evidence from Clinical Studies
4.3. Evidence from Preclinical Studies
5. SGLT2i—Effect on Serum Uric Acid Concentration: Findings in Clinical Studies
5.1. Effect of SGLT2is on Serum Uric Acid Concentration
5.2. Drug- and Dose-Dependency
5.3. SUA Reduction in Chronic Kidney Disease Patients
5.4. Factors Influencing SGLT2i Efficacy in Reducing Serum Uric Acid Level
5.5. Effect of SGLT2is on Acute Gout Events and New Antigout Drugs Commencement
5.6. SLGT2i—Novel Mechanism of Action
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Teo, Y.H.; Teo, Y.N.; Syn, N.L.; Kow, C.S.; Yoong, C.S.Y.; Tan, B.Y.Q.; Yeo, T.C.; Lee, C.H.; Lin, W.; Sia, C.H. Effects of Sodium/Glucose Cotransporter 2 (SGLT2) Inhibitors on Cardiovascular and Metabolic Outcomes in Patients Without Diabetes Mellitus: A Systematic Review and Meta-Analysis of Randomized-Controlled Trials. J. Am. Heart Assoc. 2021, 10, e019463. [Google Scholar] [CrossRef]
- Wijayanti, L.; Wahyudi, A.; Septianingrum, Y. The Correlation between Lifestyle and Increased Uric Acid Levels in the Elderly. Nurse Holist. Care 2021, 1, 48–55. [Google Scholar] [CrossRef]
- El Ridi, R.; Tallima, H. Physiological functions and pathogenic potential of uric acid: A review. J. Adv. Res. 2017, 8, 487–493. [Google Scholar] [CrossRef]
- Ruoff, G.; Edwards, N.L. Overview of Serum Uric Acid Treatment Targets in Gout: Why Less Than 6 mg/dL? Postgrad. Med. 2016, 128, 706–715. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.W.; Lin, S.Y.; Kuo, C.C.; Huang, C.C. Serum Uric Acid and Progression of Kidney Disease: A Longitudinal Analysis and Mini-Review. PLoS ONE 2017, 12, e0170393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, B.; Badve, S.V.; Hiremath, S.S.; Boudville, N.; Brown, F.G.; Cass, A.; de Zoysa, J.R.; Fassett, R.G.; Faull, R.; Harris, D.C.; et al. Effects of uric acid-lowering therapy on renal outcomes: A systematic review and meta-analysis. Nephrol. Dial. Transplant. 2014, 29, 406–413. [Google Scholar] [CrossRef] [Green Version]
- Ponticelli, C.; Podesta, M.A.; Moroni, G. Hyperuricemia as a trigger of immune response in hypertension and chronic kidney disease. Kidney Int. 2020, 98, 1149–1159. [Google Scholar] [CrossRef]
- Lin, J.D.; Chiou, W.K.; Chang, H.Y.; Liu, F.H.; Weng, H.F. Serum uric acid and leptin levels in metabolic syndrome: A quandary over the role of uric acid. Metabolism 2007, 56, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.; Zhu, W.H.; Chen, Z.W.; Dai, H.L.; Ren, J.J.; Chen, J.H.; Chen, L.Q.; Fang, L.Z. Relationship between hyperuricemia and metabolic syndrome. J. Zhejiang Univ. Sci. B. 2007, 8, 593–598. [Google Scholar] [CrossRef]
- Dehghan, A.; van Hoek, M.; Sijbrands, E.J.; Hofman, A.; Witteman, J.C. High serum uric acid as a novel risk factor for type 2 diabetes. Diabetes Care 2008, 31, 361–362. [Google Scholar] [CrossRef] [Green Version]
- Sluijs, I.; Beulens, J.W.; van der A, A.D.; Spijkerman, A.M.; Schulze, M.B.; van der Schouw, Y.T. Plasma uric acid is associated with increased risk of type 2 diabetes independent of diet and metabolic risk factors. J. Nutr. 2013, 143, 80–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Q.; Meng, X.F.; He, F.F.; Chen, S.; Su, H.; Xiong, J.; Gao, P.; Tian, X.J.; Liu, J.S.; Zhu, Z.H.; et al. High serum uric acid and increased risk of type 2 diabetes: A systemic review and meta-analysis of prospective cohort studies. PLoS ONE 2013, 8, e56864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurajoh, M.; Fukumoto, S.; Yoshida, S.; Akari, S.; Murase, T.; Nakamura, T.; Ishii, H.; Yoshida, H.; Nagata, Y.; Morioka, T.; et al. Uric acid shown to contribute to increased oxidative stress level independent of xanthine oxidoreductase activity in MedCity21 health examination registry. Sci. Rep. 2021, 11, 7378. [Google Scholar] [CrossRef]
- Yu, S.; Chen, Y.; Hou, X.; Xu, D.; Che, K.; Li, C.; Yan, S.; Wang, Y.; Wang, B. Serum Uric Acid Levels and Diabetic Peripheral Neuropathy in Type 2 Diabetes: A Systematic Review and Meta-analysis. Mol. Neurobiol. 2016, 53, 1045–1051. [Google Scholar] [CrossRef]
- Wang, L.; Hu, W.; Wang, J.; Qian, W.; Xiao, H. Low serum uric acid levels in patients with multiple sclerosis and neuromyelitis optica: An updated meta-analysis. Mult. Scler. Relat. Disord. 2016, 9, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Oppedisano, F.; Gratteri, S.; Muscoli, C.; Mollace, V. Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 2016, 213, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Meng, X.; Timofeeva, M.; Tzoulaki, I.; Tsilidis, K.K.; Ioannidis, J.P.; Campbell, H.; Theodoratou, E. Serum uric acid levels and multiple health outcomes: Umbrella review of evidence from observational studies, randomised controlled trials, and Mendelian randomisation studies. BMJ 2017, 357, j2376. [Google Scholar] [CrossRef] [Green Version]
- Moris, D.; Spartalis, M.; Spartalis, E.; Karachaliou, G.S.; Karaolanis, G.I.; Tsourouflis, G.; Tsilimigras, D.I.; Tzatzaki, E.; Theocharis, S. The role of reactive oxygen species in the pathophysiology of cardiovascular diseases and the clinical significance of myocardial redox. Ann. Transl. Med. 2017, 5, 326. [Google Scholar] [CrossRef] [Green Version]
- Patetsios, P.; Song, M.; Shutze, W.P.; Pappas, C.; Rodino, W.; Ramirez, J.A.; Panetta, T.F. Identification of uric acid and xanthine oxidase in atherosclerotic plaque. Am. J. Cardiol. 2001, 88, 188–191.A6. [Google Scholar] [CrossRef]
- Johnson, R.J.; Kang, D.H.; Feig, D.; Kivlighn, S.; Kanellis, J.; Watanabe, S.; Tuttle, K.R.; Rodriguez-Iturbe, B.; Herrera-Acosta, J.; Mazzali, M. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension 2003, 41, 1183–1190. [Google Scholar] [CrossRef] [Green Version]
- Chuang, S.Y.; Chen, J.H.; Yeh, W.T.; Wu, C.C.; Pan, W.H. Hyperuricemia and increased risk of ischemic heart disease in a large Chinese cohort. Int. J. Cardiol. 2012, 154, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Chuang, S.Y.; Chen, H.J.; Yeh, W.T.; Pan, W.H. Serum uric acid level as an independent risk factor for all-cause, cardiovascular, and ischemic stroke mortality: A Chinese cohort study. Arthritis Rheum. 2009, 61, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Hu, X.; Fan, Y.; Li, K.; Zhang, X.; Hou, W.; Tang, Z. Hyperuricemia and the risk for coronary heart disease morbidity and mortality a systematic review and dose-response meta-analysis. Sci. Rep. 2016, 6, 19520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culleton, B.F.; Larson, M.G.; Kannel, W.B.; Levy, D. Serum uric acid and risk for cardiovascular disease and death: The Framingham Heart Study. Ann. Intern. Med. 1999, 131, 7–13. [Google Scholar] [CrossRef]
- Niskanen, L.K.; Laaksonen, D.E.; Nyyssonen, K.; Alfthan, G.; Lakka, H.M.; Lakka, T.A.; Salonen, J.T. Uric acid level as a risk factor for cardiovascular and all-cause mortality in middle-aged men: A prospective cohort study. Arch. Intern. Med. 2004, 164, 1546–1551. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, J.G.; Juzwishin, K.D.; Eiriksdottir, G.; Gudnason, V.; Danesh, J. Serum uric acid and coronary heart disease in 9458 incident cases and 155,084 controls: Prospective study and meta-analysis. PLoS Med. 2005, 2, e76. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.S.; Hawkey, C.J.; Ford, I.; Greenlaw, N.; Pigazzani, F.; Rogers, A.; Struthers, A.D.; Begg, A.G.; Wei, L.; Avery, A.J.; et al. Allopurinol versus usual care in UK patients with ischaemic heart disease (ALL-HEART): A multicentre, prospective, randomised, open-label, blinded-endpoint trial. Lancet 2022, 400, 1195–1205. [Google Scholar] [CrossRef]
- Leyva, F.; Anker, S.; Swan, J.W.; Godsland, I.F.; Wingrove, C.S.; Chua, T.P.; Stevenson, J.C.; Coats, A.J. Serum uric acid as an index of impaired oxidative metabolism in chronic heart failure. Eur. Heart J. 1997, 18, 858–865. [Google Scholar] [CrossRef] [Green Version]
- Leyva, F.; Chua, T.P.; Anker, S.D.; Coats, A.J. Uric acid in chronic heart failure: A measure of the anaerobic threshold. Metabolism 1998, 47, 1156–1159. [Google Scholar] [CrossRef]
- Huang, H.; Huang, B.; Li, Y.; Huang, Y.; Li, J.; Yao, H.; Jing, X.; Chen, J.; Wang, J. Uric acid and risk of heart failure: A systematic review and meta-analysis. Eur. J. Heart Fail. 2014, 16, 15–24. [Google Scholar] [CrossRef]
- Cicoira, M.; Zanolla, L.; Rossi, A.; Golia, G.; Franceschini, L.; Brighetti, G.; Zeni, P.; Zardini, P. Elevated serum uric acid levels are associated with diastolic dysfunction in patients with dilated cardiomyopathy. Am. Heart J. 2002, 143, 1107–1111. [Google Scholar] [CrossRef]
- Feig, D.I.; Kang, D.H.; Johnson, R.J. Uric acid and cardiovascular risk. N. Engl. J. Med. 2008, 359, 1811–1821. [Google Scholar] [CrossRef]
- Beattie, C.J.; Fulton, R.L.; Higgins, P.; Padmanabhan, S.; McCallum, L.; Walters, M.R.; Dominiczak, A.F.; Touyz, R.M.; Dawson, J. Allopurinol initiation and change in blood pressure in older adults with hypertension. Hypertension 2014, 64, 1102–1107. [Google Scholar] [CrossRef] [Green Version]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [Green Version]
- Tonneijck, L.; Muskiet, M.H.; Smits, M.M.; van Bommel, E.J.; Heerspink, H.J.; van Raalte, D.H.; Joles, J.A. Glomerular Hyperfiltration in Diabetes: Mechanisms, Clinical Significance, and Treatment. J. Am. Soc. Nephrol. 2017, 28, 1023–1039. [Google Scholar] [CrossRef] [Green Version]
- van Bommel, E.J.M.; Muskiet, M.H.A.; van Baar, M.J.B.; Tonneijck, L.; Smits, M.M.; Emanuel, A.L.; Bozovic, A.; Danser, A.H.J.; Geurts, F.; Hoorn, E.J.; et al. The renal hemodynamic effects of the SGLT2 inhibitor dapagliflozin are caused by post-glomerular vasodilatation rather than pre-glomerular vasoconstriction in metformin-treated patients with type 2 diabetes in the randomized, double-blind RED trial. Kidney Int. 2020, 97, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, M.; Yang, G.K.; Perkins, B.A.; Soleymanlou, N.; Lytvyn, Y.; von Eynatten, M.; Woerle, H.J.; Johansen, O.E.; Broedl, U.C.; Hach, T.; et al. Characterisation of glomerular haemodynamic responses to SGLT2 inhibition in patients with type 1 diabetes and renal hyperfiltration. Diabetologia 2014, 57, 2599–2602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2021, 83, 503–528. [Google Scholar] [CrossRef] [PubMed]
- Cassis, P.; Locatelli, M.; Cerullo, D.; Corna, D.; Buelli, S.; Zanchi, C.; Villa, S.; Morigi, M.; Remuzzi, G.; Benigni, A.; et al. SGLT2 inhibitor dapagliflozin limits podocyte damage in proteinuric nondiabetic nephropathy. JCI Insight 2018, 3, e98720. [Google Scholar] [CrossRef] [Green Version]
- Novikov, A.; Vallon, V. Sodium glucose cotransporter 2 inhibition in the diabetic kidney: An update. Curr. Opin. Nephrol. Hypertens. 2016, 25, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Silva Dos Santos, D.; Polidoro, J.Z.; Borges-Junior, F.A.; Girardi, A.C.C. Cardioprotection conferred by sodium-glucose cotransporter 2 inhibitors: A renal proximal tubule perspective. Am. J. Physiol. Cell. Physiol. 2020, 318, C328–C336. [Google Scholar] [CrossRef] [PubMed]
- Perico, N.; Ruggenenti, P.; Remuzzi, G. ACE and SGLT2 inhibitors: The future for non-diabetic and diabetic proteinuric renal disease. Curr. Opin. Pharmacol. 2017, 33, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Levi, J.; Luo, Y.; Myakala, K.; Herman-Edelstein, M.; Qiu, L.; Wang, D.; Peng, Y.; Grenz, A.; Lucia, S.; et al. SGLT2 Protein Expression Is Increased in Human Diabetic Nephropathy: SGLT2 Protein Inhibition Decreases Renal Lipid Accumulation, Inflammation, and the Development of Nephropathy in Diabetic Mice. J. Biol. Chem. 2017, 292, 5335–5348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; 636 Anand, I.S.; Belohlavek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 637, 1995–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, S.; Zhu, P.; Hu, S.; Chen, Y.; Ren, J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018, 15, 335–346. [Google Scholar] [CrossRef]
- Lazarte, J.; Kanagalingam, T.; Hegele, R.A. Lipid effects of sodium-glucose cotransporter 2 inhibitors. Curr. Opin. Lipidol. 2021, 32, 183–190. [Google Scholar] [CrossRef]
- Reed, J.W. Impact of sodium-glucose cotransporter 2 inhibitors on blood pressure. Vasc. Health Risk Manag. 2016, 12, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Konishi, Y.; Morikawa, T.; Zhang, Y.; Kitabayashi, C.; Kobara, H.; Masaki, T.; Nakano, D.; Hitomi, H.; Kobori, H.; et al. Effect of a SGLT2 inhibitor on the systemic and intrarenal renin-angiotensin system in subtotally nephrectomized rats. J. Pharmacol. Sci. 2018, 137, 220–223. [Google Scholar] [CrossRef]
- Mori, I.; Ishizuka, T. Effects of SGLT2 Inhibitors on Renin-Aldosterone System for One Month and Six Months in Type 2 Diabetes. Diabetes 2018, 67, 1196. [Google Scholar] [CrossRef]
- Hou, Y.C.; Zheng, C.M.; Yen, T.H.; Lu, K.C. Molecular Mechanisms of SGLT2 Inhibitor on Cardiorenal Protection. Int. J. Mol. Sci. 2020, 21, 7833. [Google Scholar] [CrossRef]
- Schlaich, M.; Straznicky, N.; Lambert, E.; Lambert, G. Metabolic syndrome: A sympathetic disease? Lancet Diabetes Endocrinol. 2015, 3, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Kalil, G.Z.; Haynes, W.G. Sympathetic nervous system in obesity-related hypertension: Mechanisms and clinical implications. Hypertens. Res. 2012, 35, 4–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuo, K.; Rakugi, H.; Ogihara, T.; Esler, M.D.; Lambert, G.W. Cardiovascular and renal complications of type 2 diabetes in obesity: Role of sympathetic nerve activity and insulin resistance. Curr. Diabetes Rev. 2010, 6, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Sano, M. A new class of drugs for heart failure: SGLT2 inhibitors reduce sympathetic overactivity. J. Cardiol. 2018, 71, 471–476. [Google Scholar] [CrossRef] [Green Version]
- DiBona, G.F. Sympathetic nervous system and the kidney in hypertension. Curr. Opin. Nephrol. Hypertens. 2002, 11, 197–200. [Google Scholar] [CrossRef]
- Rahman, A.; Fujisawa, Y.; Nakano, D.; Hitomi, H.; Nishiyama, A. Effect of a selective SGLT2 inhibitor, luseogliflozin, on circadian rhythm of sympathetic nervous function and locomotor activities in metabolic syndrome rats. Clin. Exp. Pharmacol. Physiol. 2017, 44, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef]
- Xu, L.; Ota, T. Emerging roles of SGLT2 inhibitors in obesity and insulin resistance: Focus on fat browning and macrophage polarization. Adipocyte 2018, 7, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Okauchi, S.; Shimoda, M.; Obata, A.; Kimura, T.; Hirukawa, H.; Kohara, K.; Mune, T.; Kaku, K.; Kaneto, H. Protective effects of SGLT2 inhibitor luseogliflozin on pancreatic beta-cells in obese type 2 diabetic db/db mice. Biochem. Biophys. Res. Commun. 2016, 470, 772–782. [Google Scholar] [CrossRef]
- Kaneto, H.; Obata, A.; Kimura, T.; Shimoda, M.; Kinoshita, T.; Matsuoka, T.A.; Kaku, K. Unexpected Pleiotropic Effects of SGLT2 Inhibitors: Pearls and Pitfalls of This Novel Antidiabetic Class. Int. J. Mol. Sci. 2021, 22, 62. [Google Scholar] [CrossRef] [PubMed]
- Androutsakos, T.; Nasiri-Ansari, N.; Bakasis, A.D.; Kyrou, I.; Efstathopoulos, E.; Randeva, H.S.; Kassi, E. SGLT-2 Inhibitors in NAFLD: Expanding Their Role beyond Diabetes and Cardioprotection. Int. J. Mol. Sci. 2022, 23, 3107. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Atkin, S.L.; Butler, A.E.; Sahebkar, A. Sodium-glucose cotransporter inhibitors and oxidative stress: An update. J. Cell. Physiol. 2019, 234, 3231–3237. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [Green Version]
- Prattichizzo, F.; De Nigris, V.; Micheloni, S.; La Sala, L.; Ceriello, A. Increases in circulating levels of ketone bodies and cardiovascular protection with SGLT2 inhibitors: Is low-grade inflammation the neglected component? Diabetes Obes. Metab. 2018, 20, 2515–2522. [Google Scholar] [CrossRef]
- Tan, S.A.; Tan, L. Empagliflozin and Canagliflozin Attenuate Inflammatory Cytokines Interferon-, Tumor Necrosis Factor-Interleukin-6: Possible Mechanism of Decreasing Cardiovascular Risk in Diabetes Mellitus. J. Am. Coll. Cardiol. 2018, 71, A1830. [Google Scholar] [CrossRef]
- Maruyama, T.; Takashima, H.; Oguma, H.; Nakamura, Y.; Ohno, M.; Utsunomiya, K.; Furukawa, T.; Tei, R.; Abe, M. Canagliflozin Improves Erythropoiesis in Diabetes Patients with Anemia of Chronic Kidney Disease. Diabetes Technol. Ther. 2019, 21, 713–720. [Google Scholar] [CrossRef]
- Bailey, C.J. Uric acid and the cardio-renal effects of SGLT2 inhibitors. Diabetes Obes. Metab. 2019, 21, 1291–1298. [Google Scholar] [CrossRef] [Green Version]
- Chino, Y.; Samukawa, Y.; Sakai, S.; Nakai, Y.; Yamaguchi, J.; Nakanishi, T.; Tamai, I. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm. Drug. Dispos. 2014, 35, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.H.; Chang, Y.P.; Li, T.; Han, F.; Li, C.J.; Li, X.Y.; Xue, M.; Cheng, Y.; Meng, Z.Y.; Han, Z.; et al. Empagliflozin Attenuates Hyperuricemia by Upregulation of ABCG2 via AMPK/AKT/CREB Signaling Pathway in Type 2 Diabetic Mice. Int. J. Biol. Sci. 2020, 16, 529–542. [Google Scholar] [CrossRef]
- Ng, H.Y.; Leung, F.F.; Kuo, W.H.; Lee, W.C.; Lee, C.T. Dapagliflozin and xanthine oxidase inhibitors improve insulin resistance and modulate renal glucose and urate transport in metabolic syndrome. Clin. Exp. Pharmacol. Physiol. 2021, 48, 1603–1612. [Google Scholar] [CrossRef]
- Novikov, A.; Fu, Y.; Huang, W.; Freeman, B.; Patel, R.; van Ginkel, C.; Koepsell, H.; Busslinger, M.; Onishi, A.; Nespoux, J.; et al. SGLT2 inhibition and renal urate excretion: Role of luminal glucose, GLUT9, and URAT1. Am. J. Physiol. Renal Physiol. 2019, 316, F173–F185. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, M.; Oba, K.; Kaku, K.; Suganami, H.; Yoshida, A.; Fukunaka, Y.; Jutabha, P.; Morita, A.; Otani, N.; Hayashi, K.; et al. Uric acid lowering in relation to HbA1c reductions with the SGLT2 inhibitor tofogliflozin. Diabetes Obes. Metab. 2018, 20, 1061–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Xu, L.; Tian, D.; Xia, P.; Zheng, H.; Wang, L.; Chen, L. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: A meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2018, 20, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Lytvyn, Y.; Skrtic, M.; Yang, G.K.; Yip, P.M.; Perkins, B.A.; Cherney, D.Z. Glycosuria-mediated urinary uric acid excretion in patients with uncomplicated type 1 diabetes mellitus. Am. J. Physiol. Ren. Physiol. 2015, 308, F77–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoki, D.; Shibata, S.; Kuribayashi-Okuma, E.; Xu, N.; Ishizawa, K.; Hosoyamada, M.; Uchida, S. Insulin stimulates uric acid reabsorption via regulating urate transporter 1 and ATP-binding cassette subfamily G member 2. Am. J. Physiol. Renal Physiol. 2017, 313, F826–F834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. A novel approach to control hyperglycemia in type 2 diabetes: Sodium glucose co-transport (SGLT) inhibitors: Systematic review and meta-analysis of randomized trials. Ann. Med. 2012, 44, 375–393. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, L.; Wu, B.; Song, H.; An, Z.; Li, S. Dapagliflozin treatment for type 2 diabetes: A systematic review and meta-analysis of randomized controlled trials. Diabetes Metab. Res. Rev. 2014, 30, 204–221. [Google Scholar] [CrossRef]
- Xin, Y.; Guo, Y.; Li, Y.; Ma, Y.; Li, L.; Jiang, H. Effects of sodium glucose cotransporter-2 inhibitors on serum uric acid in type 2 diabetes mellitus: A systematic review with an indirect comparison meta-analysis. Saudi J. Biol. Sci. 2019, 26, 421–426. [Google Scholar] [CrossRef]
- Zhao, D.; Liu, H.; Dong, P. Empagliflozin reduces blood pressure and uric acid in patients with type 2 diabetes mellitus: A systematic review and meta-analysis. J. Hum. Hypertens. 2019, 33, 327–339. [Google Scholar] [CrossRef]
- Wu, B.; Zheng, H.; Gu, J.; Guo, Y.; Liu, Y.; Wang, Y.; Chen, F.; Yang, A.; Wang, J.; Wang, H.; et al. Effects of sodium-glucose cotransporter 2 inhibitors in addition to insulin therapy on cardiovascular risk factors in type 2 diabetes patients: A meta-analysis of randomized controlled trials. J. Diabetes Investig. 2019, 10, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yang, Y.; Hu, X.; Jia, X.; Liu, H.; Wei, M.; Lyu, Z. Effects of sodium-glucose cotransporter 2 inhibitors on serum uric acid in patients with type 2 diabetes mellitus: A systematic review and network meta-analysis. Diabetes Obes. Metab. 2022, 24, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Akbari, A.; Rafiee, M.; Sathyapalan, T.; Sahebkar, A. Impacts of Sodium/Glucose Cotransporter-2 Inhibitors on Circulating Uric Acid Concentrations: A Systematic Review and Meta-Analysis. J. Diabetes Res. 2022, 2022, 7520632. [Google Scholar] [CrossRef] [PubMed]
- Yip, A.S.Y.; Leong, S.; Teo, Y.H.; Teo, Y.N.; Syn, N.L.X.; See, R.M.; Wee, C.F.; Chong, E.Y.; Lee, C.H.; Chan, M.Y.; et al. Effect of sodium-glucose cotransporter-2 (SGLT2) inhibitors on serum urate levels in patients with and without diabetes: A systematic review and meta-regression of 43 randomized controlled trials. Ther. Adv. Chronic Dis. 2022, 13, 20406223221083509. [Google Scholar] [CrossRef]
- Wang, S.; Yuan, T.; Song, S.; Duo, Y.; Zhao, T.; Gao, J.; Fu, Y.; Dong, Y.; Zhao, W. Medium- and Long-Term Effects of Dapagliflozin on Serum Uric Acid Level in Patients with Type 2 Diabetes: A Real-World Study. J. Pers. Med. 2022, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Liu, S.; Dong, Y.; Fu, Y.; Tang, Y.; Zhao, W. Effects of dapagliflozin on serum and urinary uric acid levels in patients with type 2 diabetes: A prospective pilot trial. Diabetol. Metab. Syndr. 2020, 12, 92. [Google Scholar] [CrossRef]
- Chino, Y.; Kuwabara, M.; Hisatome, I. Factors Influencing Change in Serum Uric Acid After Administration of the Sodium- 749 Glucose Cotransporter 2 Inhibitor Luseogliflozin in Patients with Type 2 Diabetes Mellitus. J. Clin. Pharmacol. 2022, 62, 366–375. [Google Scholar] [CrossRef]
- Doehner, W.; Anker, S.D.; Butler, J.; Zannad, F.; Filippatos, G.; Ferreira, J.P.; Salsali, A.; Kaempfer, C.; Brueckmann, M.; Pocock, S.J.; et al. Uric acid and sodium-glucose cotransporter-2 inhibition with empagliflozin in heart failure with reduced ejection fraction: The EMPEROR-reduced trial. Eur. Heart J. 2022, 43, 3435–3446. [Google Scholar] [CrossRef]
- Li, J.; Badve, S.V.; Zhou, Z.; Rodgers, A.; Day, R.; Oh, R.; Lee, M.; Perkovic, V.; de Zeeuw, D.; Mahaffey, K.W.; et al. The effects of canagliflozin on gout in type 2 diabetes: A post-hoc analysis of the CANVAS Program. Lancet Rheumatol. 2019, 1, e220–e228. [Google Scholar] [CrossRef]
- Zanchi, A.; Burnier, M.; Muller, M.E.; Ghajarzadeh-Wurzner, A.; Maillard, M.; Loncle, N.; Milani, B.; Dufour, N.; Bonny, O.; Pruijm, M. Acute and Chronic Effects of SGLT2 Inhibitor Empagliflozin on Renal Oxygenation and Blood Pressure Control in Nondiabetic Normotensive Subjects: A Randomized, Placebo-Controlled Trial. J. Am. Heart Assoc. 2020, 9, e016173. [Google Scholar] [CrossRef]
- Okada, K.; Hoshide, S.; Kato, M.; Kanegae, H.; Ishibashi, S.; Kario, K. Safety and efficacy of empagliflozin in elderly Japanese patients with type 2 diabetes mellitus: A post hoc analysis of data from the SACRA study. J. Clin. Hypertens. 2021, 23, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Koshizaka, M.; Ishikawa, K.; Ishibashi, R.; Takahashi, S.; Sakamoto, K.; Yokoh, H.; Baba, Y.; Ide, S.; Ide, K.; Ishikawa, T.; et al. Comparison of Visceral Fat Reduction by Ipragliflozin and Metformin in Elderly Type 2 Diabetes Patients: Sub-Analysis of a Randomized-Controlled Study. Diabetes Ther. 2021, 12, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.; Filippatos, G.; Siddiqi, T.J.; Ferreira, J.P.; Brueckmann, M.; Bocchi, E.; Bohm, M.; Chopra, V.K.; Giannetti, N.; Iwata, T.; et al. Effects of Empagliflozin in Women and Men with Heart Failure and Preserved Ejection Fraction. Circulation 2022, 146, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Gunhan, H.G.; Imre, E.; Erel, P.; Ustay, O. Empagliflozin Is More Effective in Reducing Microalbuminuria and Alt Levels Compared with Dapagliflozin: Real Life Experience. Acta Endocrinol. 2020, 16, 59–67. [Google Scholar] [CrossRef]
- Grempler, R.; Thomas, L.; Eckhardt, M.; Himmelsbach, F.; Sauer, A.; Sharp, D.E.; Bakker, R.A.; Mark, M.; Klein, T.; Eickelmann, P. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: Characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes. Metab. 2012, 14, 83–90. [Google Scholar] [CrossRef]
- Sasaki, T.; Seino, Y.; Fukatsu, A.; Ubukata, M.; Sakai, S.; Samukawa, Y. Pharmacokinetics, Pharmacodynamics, and Safety of Luseogliflozin in Japanese Patients with Type 2 Diabetes Mellitus: A Randomized, Single-blind, Placebo-controlled Trial. Adv. Ther. 2015, 32, 319–340. [Google Scholar] [CrossRef] [Green Version]
- Yale, J.F.; Bakris, G.; Cariou, B.; Nieto, J.; David-Neto, E.; Yue, D.; Wajs, E.; Figueroa, K.; Jiang, J.; Law, G.; et al. Efficacy and safety of canagliflozin over 52 weeks in patients with type 2 diabetes mellitus and chronic kidney disease. Diabetes Obes. Metab. 2014, 16, 1016–1027. [Google Scholar] [CrossRef]
- Barnett, A.H.; Mithal, A.; Manassie, J.; Jones, R.; Rattunde, H.; Woerle, H.J.; Broedl, U.C.; EMPA-REG RENAL Trial Investigators. Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: A randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014, 2, 369–384. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefansson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- Rossing, P.; Caramori, M.L.; Chan, J.C.N.; Heerspink, H.J.L.; Hurst, C.; Khunti, K.; Liew, A.; Michos, E.D.; Navaneethan, S.D.; Olowu, W.A.; et al. Executive summary of the KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease: An update based on rapidly emerging new evidence. Kidney Int. 2022, 102, 990–999. [Google Scholar] [CrossRef]
- McDowell, K.; Welsh, P.; Docherty, K.F.; Morrow, D.A.; Jhund, P.S.; de Boer, R.A.; O’Meara, E.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; et al. Dapagliflozin reduces uric acid concentration, an independent predictor of adverse outcomes in DAPA-HF. Eur. J. Heart Fail. 2022, 24, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Gouda, M.; Arakawa, K.; Inagaki, M.; Ushirogawa, Y. Effect of sodium-glucose cotransporter 2 inhibitor medication on new prescriptions of antihypertensives, antigout/antihyperuricemics and antidyslipidemics in Japan: Analysis using the JMDC Claims Database. J. Diabetes Investig. 2022, 13, 1842–1851. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Pal, R.; Mukhopadhyay, S. Can SGLT2 inhibitors prevent incident gout? A systematic review and meta-analysis. Acta Diabetol. 2022, 59, 783–791. [Google Scholar] [CrossRef]
- Stack, A.G.; Han, D.; Goldwater, R.; Johansson, S.; Dronamraju, N.; Oscarsson, J.; Johnsson, E.; Parkinson, J.; Erlandsson, F. Dapagliflozin Added to Verinurad Plus Febuxostat Further Reduces Serum Uric Acid in Hyperuricemia: The QUARTZ Study. J. Clin. Endocrinol. Metab. 2021, 106, e2347–e2356. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, C.; Dicembrini, I.; Nreu, B.; Mannucci, E.; Monami, M. Nephrolithiasis and sodium-glucose co-transporter-2 (SGLT-2) inhibitors: A meta-analysis of randomized controlled trials. Diabetes Res. Clin. Pract. 2019, 155, 107808. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Author | Year | No. of Trials | No. of Individuals | Population | Drug | Uric Acid (µmol/L) | I2 |
|---|---|---|---|---|---|---|---|
| Musso [77] | 2012 | 7 | 2943 | T2D | dapagliflozin | −41.50 (−47.22, −35.79) a | 50% |
| Zhang [78] | 2014 | 10 | 3464 | T2D | dapagliflozin | −36.17 (−40.99, −31.36) a | 64% |
| Zhao [74] | 2018 | 62 | 34,941 | T2D | total | −37.73 (−40.51, −34.95) a | 73.5% |
| empagliflozin | −45.83 (−53.03, −38.63) a | 0% | |||||
| dapagliflozin | −36.99 (−41.73, −32.25) a | 65.4% | |||||
| canagliflozin | −41.22 (−45.03, −37.42) a | 81.4% | |||||
| ipragliflozin | −17.40 (−23.78, −11.02) a | 12.5% | |||||
| luseogliflozin | −28.20 (−34.73, −21.67) a | 11.6% | |||||
| tofogliflozin | −21.48 (−35.15, −7.81) a | 0% | |||||
| Xin [79] | 2019 | 31 | 13,650 | T2D | canagliflozin | −37.02 (38.41, −35.63) a | 80% |
| dapagliflozin | −38.05 (−44.47, −31.62) a | 99% | |||||
| empagliflozin | −42.07 (−46.27, −37.86) a | 67% | |||||
| tofogliflozin | −18.97 (−28.79, −9.16) a | 7% | |||||
| ipragliflozin | −19.75 (−28.17, −11.34) a | 0% | |||||
| Zhao [80] | 2019 | 12 | 5781 | T2D | Empagliflozin c | −36.59 (−46.22, −26.96) a | 65% |
| Empagliflozin d | −43.55 (−52.40, −34.70) a | 65% | |||||
| Wu [81] | 2019 | 10 | 5159 | T2D | total | −26.16 (−42.14, −10.17) a | 80% |
| Hu [82] | 2022 | 19 | 4218 | T2D | total | −0.965 (−1.029, −0.901) a,b | 98.7% |
| empagliflozin | −0.710 (−0.832, −0.587) a,b | 0% | |||||
| dapagliflozin | −2.787 (−2.965, −2.610) a,b | 98.9% | |||||
| canagliflozin | −0.503 (−0.639, −0.366) a,b | 0% | |||||
| ipragliflozin | −0.294 (−0.438, −0.151) a,b | 87.6% | |||||
| luseogliflozin | −6.916 (−7.288, −6.544) a,b | 97.9% | |||||
| tofogliflozin | −0.184 (−0.357, −0.011) a,b | 86.3% | |||||
| Akbari [83] | 2022 | 55 | 36,215 | T2D | total | −34.07 (−37.00, −31.14) a | 78.8% |
| empagliflozin | −40.98 (−47.63, −34.32) a | 84.9% | |||||
| dapagliflozin | −35.17 (−39.68, −30.66) a | 73.9% | |||||
| canagliflozin | −36.27 (−41.62, −30.93) a | 66.5% | |||||
| luseogliflozin | −24.269 (−33.31, −15.22) a | 66.3% | |||||
| tofogliflozin | −19.47 (−27.40, −11.55) a | 0% | |||||
| ipragliflozin | −18.85 (−27.20, −10.49) a | 59% | |||||
| Yip [84] | 2022 | 43 | 31,921 | total | total | −33.03 (−37.38, −28.69) a | 92% |
| 3 | 597 | luseogliflozin | −47.73 (−79.50, −15.96) a | 94% | |||
| 7 | 4002 | canagliflozin | −36.62 (−42.67, −30.56) a | 61% | |||
| 16 | 17,653 | empagliflozin | −35.19 (−42.61, −27.78) a | 96% | |||
| 15 | 5036 | dapagliflozin | −30.32 (−36.20, −24.43) a | 67% | |||
| 2 | 702 | ipragliflozin | −20.37 (−29.17, −11.56) a | 72% | |||
| 4 | 198 | without T2D | total | −91.38 (−126.53, −56.24) a | 80% | ||
| 39 | 31,723 | T2D | total | −31.48 (−37.35, −25.60) a | 92% | ||
| 8 | CKD | total | −8.12 (−22.17, 5.94), p = 0.26 | 69% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kochanowska, A.; Rusztyn, P.; Szczerkowska, K.; Surma, S.; Gąsecka, A.; Jaguszewski, M.J.; Szarpak, Ł.; Filipiak, K.J. Sodium–Glucose Cotransporter 2 Inhibitors to Decrease the Uric Acid Concentration—A Novel Mechanism of Action. J. Cardiovasc. Dev. Dis. 2023, 10, 268. https://doi.org/10.3390/jcdd10070268
Kochanowska A, Rusztyn P, Szczerkowska K, Surma S, Gąsecka A, Jaguszewski MJ, Szarpak Ł, Filipiak KJ. Sodium–Glucose Cotransporter 2 Inhibitors to Decrease the Uric Acid Concentration—A Novel Mechanism of Action. Journal of Cardiovascular Development and Disease. 2023; 10(7):268. https://doi.org/10.3390/jcdd10070268
Chicago/Turabian StyleKochanowska, Anna, Przemysław Rusztyn, Karolina Szczerkowska, Stanisław Surma, Aleksandra Gąsecka, Miłosz J. Jaguszewski, Łukasz Szarpak, and Krzysztof J. Filipiak. 2023. "Sodium–Glucose Cotransporter 2 Inhibitors to Decrease the Uric Acid Concentration—A Novel Mechanism of Action" Journal of Cardiovascular Development and Disease 10, no. 7: 268. https://doi.org/10.3390/jcdd10070268
APA StyleKochanowska, A., Rusztyn, P., Szczerkowska, K., Surma, S., Gąsecka, A., Jaguszewski, M. J., Szarpak, Ł., & Filipiak, K. J. (2023). Sodium–Glucose Cotransporter 2 Inhibitors to Decrease the Uric Acid Concentration—A Novel Mechanism of Action. Journal of Cardiovascular Development and Disease, 10(7), 268. https://doi.org/10.3390/jcdd10070268