
Pulmonary Vasodilator Therapy Is Associated with Decreased Mortality in Patients with Chronic Lung Disease and Severe Pulmonary Hypertension


Abstract
:1. Introduction
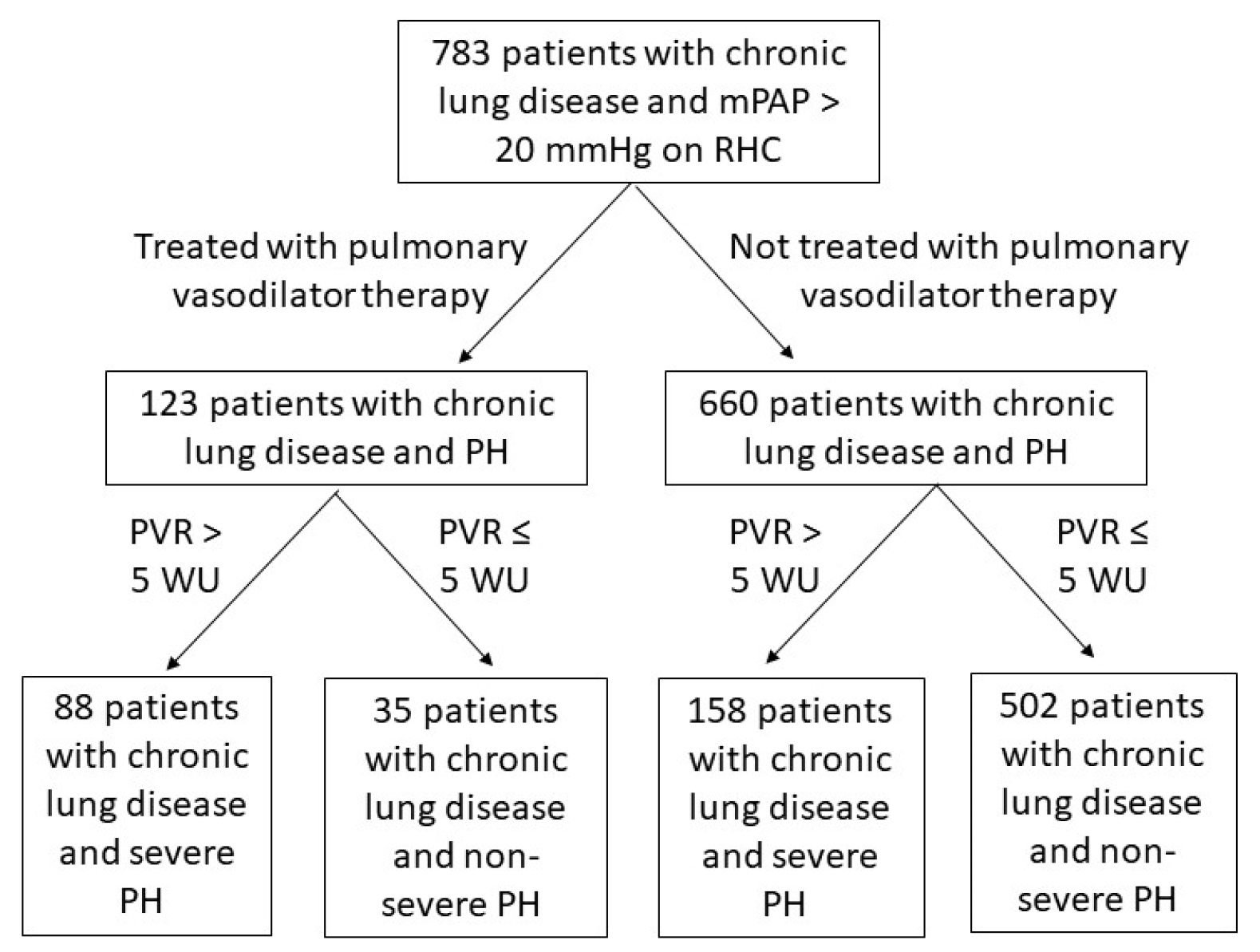
2. Materials and Methods
2.1. Study Design and Definitions
2.2. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weitzenblum, E.; Hirth, C.; Ducolone, A.; Mirhom, R.; Rasaholinjanahary, J.; Ehrhart, M. Prognostic value of pulmonary artery pressure in chronic obstructive pulmonary disease. Thorax 1981, 36, 752–758. [Google Scholar] [CrossRef]
- Shorr, A.F.; Helman, D.L.; Davies, D.B.; Nathan, S.D. Pulmonary hypertension in advanced sarcoidosis: Epidemiology and clinical characteristics. Eur. Respir. J. 2005, 25, 783–788. [Google Scholar] [CrossRef]
- Lettieri, C.J.; Nathan, S.D.; Barnett, S.D.; Ahmad, S.; Shorr, A.F. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest 2006, 129, 746–752. [Google Scholar] [CrossRef]
- Kimura, M.; Taniguchi, H.; Kondoh, Y.; Kimura, T.; Kataoka, K.; Nishiyama, O.; Aso, H.; Sakamoto, K.; Hasegawa, Y. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Respiration 2013, 85, 456–463. [Google Scholar] [CrossRef]
- Prins, K.W.; Duval, S.; Markowitz, J.; Pritzker, M.; Thenappan, T. Chronic use of PAH-specific therapy in World Health Organization Group III Pulmonary Hypertension: A systematic review and meta-analysis. Pulm. Circ. 2017, 7, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Selimovic, N.; Andersson, B.; Bergh, C.H.; Mårtensson, G.; Nilsson, F.; Bech-Hanssen, O.; Rundqvist, B. Pulmonary hemodynamics as predictors of mortality in patients awaiting lung transplantation. Transpl. Int. 2008, 21, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Zeder, K.; Avian, A.; Bachmaier, G.; Douschan, P.; Foris, V.; Sassmann, T.; Troester, N.; Brcic, L.; Fuchsjaeger, M.; Marsh, L.M.; et al. Elevated pulmonary vascular resistance predicts mortality in COPD patients. Eur. Respir. J. 2021, 58, 2100944. [Google Scholar] [CrossRef] [PubMed]
- Olsson, K.M.; Hoeper, M.M.; Pausch, C.; Grünig, E.; Huscher, D.; Pittrow, D.; Rosenkranz, S.; Gall, H. Pulmonary vascular resistance predicts mortality in patients with pulmonary hypertension associated with interstitial lung disease: Results from the COMPERA registry. Eur. Respir. J. 2021, 58, 2101483. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. G. Ital. Cardiol. 2023, 24, 1e–116e. [Google Scholar] [CrossRef] [PubMed]
- Gayen, S.K.; Baughman, R.P.; Nathan, S.D.; Wells, A.U.; Kouranos, V.; Alhamad, E.H.; Culver, D.A.; Barney, J.; Carmoma, E.M.; Cordova, F.C.; et al. Pulmonary hemodynamics and transplant-free survival in sarcoidosis-associated pulmonary hypertension: Results from an international registry. Pulm. Circ. 2023, 13, e12297. [Google Scholar] [CrossRef]
- Gayen, S.; Ansari, S.; Lashari, B.H.; Zhao, H.; Criner, G.J.; Gupta, R.; James Mamary, A. Pulmonary vasodilator therapy in sarcoidosis-associated pulmonary hypertension may decrease lung function decline and mortality. Pulm. Circ. 2023, 13, e12245. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Behr, J.; Collard, H.R.; Cottin, V.; Hoeper, M.M.; Martinez, F.J.; Corte, T.J.; Keogh, A.M.; Leuchte, H.; Mogulkoc, N.; et al. Riociguat for idiopathic interstitial pneumonia-associated pulmonary hypertension (RISE-IIP): A randomised, placebo-controlled phase 2b study. Lancet Respir. Med. 2019, 7, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Waxman, A.; Restrepo-Jaramillo, R.; Thenappan, T.; Ravichandran, A.; Engel, P.; Bajwa, A.; Allen, R.; Feldman, J.; Argula, R.; Smith, P.; et al. Inhaled Treprostinil in Pulmonary Hypertension Due to Interstitial Lung Disease. N. Engl. J. Med. 2021, 384, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Johri, S.; Joly, J.M.; King, C.S.; Raina, A.; McEvoy, C.A.; Lee, D.; Shen, E.; Smith, P.; Deng, C.; et al. Survival analysis from the INCREASE study in PH-ILD: Evaluating the impact of treatment crossover on overall mortality. Thorax 2023, 78, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Elkhapery, A.; Hammami, M.B.; Sulica, R.; Boppana, H.; Abdalla, Z.; Iyer, C.; Taifour, H.; Niu, C.; Deshwal, H. Pulmonary Vasodilator Therapy in Severe Pulmonary Hypertension Due to Chronic Obstructive Pulmonary Disease (Severe PH-COPD): A Systematic Review and Meta-Analysis. J. Cardiovasc. Dev. Dis. 2023, 10, 498. [Google Scholar] [CrossRef] [PubMed]
- Stadler, S.; Mergenthaler, N.; Lange, T.J. The prognostic value of DLCO and pulmonary blood flow in patients with pulmonary hypertension. Pulm. Circ. 2019, 9, 1–19. [Google Scholar] [CrossRef]
- Shlobin, O.A.; Kouranos, V.; Barnett, S.D.; Alhamad, E.H.; Culver, D.A.; Barney, J.; Cordova, F.C.; Carmona, E.M.; Scholand, M.B.; Wijsenbeek, M.; et al. Physiological predictors of survival in patients with sarcoidosis-associated pulmonary hypertension: Results from an international registry. Eur. Respir. J. 2020, 55, 1901747. [Google Scholar] [CrossRef] [PubMed]
- Alhamad, E.H.; Cal, J.G.; Alrajhi, N.N.; Alharbi, W.M. Predictors of Mortality in Patients with Interstitial Lung Disease-Associated Pulmonary Hypertension. J. Clin. Med. 2020, 9, 3828. [Google Scholar] [CrossRef]
- Galié, N.; Manes, A.; Negro, L.; Palazzini, M.; Bacchi-Reggiani, M.L.; Branzi, A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur. Heart J. 2009, 30, 394–403. [Google Scholar] [CrossRef]
- Barst, R.J.; Rubin, L.J.; Long, W.A.; McGoon, M.D.; Rich, S.; Badesch, D.B.; Groves, B.M.; Tapson, V.F.; Bourge, R.C.; Brundage, B.H.; et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N. Engl. J. Med. 1996, 334, 296–301. [Google Scholar] [CrossRef]
- McLaughlin, V.V.; Sitbon, O.; Badesch, D.B.; Barst, R.J.; Black, C.; Galié, N.; Rainisio, M.; Simonneau, G.; Rubin, L.J. Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur. Respir. J. 2005, 25, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galié, N.; Ghofrani, H.A.; Jansa, P.; Jing, Z.C.; Le Brun, F.O.; Mehta, S.; et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Chaouat, A.; Bugnet, A.S.; Kadaoui, N.; Schott, R.; Enache, I.; Ducoloné, A.; Ehrhart, M.; Kessler, R.; Weitzenblum, E. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2005, 172, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Shorr, A.F.; Wainright, J.L.; Cors, C.S.; Lettieri, C.J.; Nathan, S.D. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur. Respir. J. 2007, 30, 715–721. [Google Scholar] [CrossRef] [PubMed]
- King, C.S.; Shlobin, O.A. The Trouble with Group 3 Pulmonary Hypertension in Interstitial Lung Disease: Dilemmas in Diagnosis and the Conundrum of Treatment. Chest 2020, 158, 1651–1664. [Google Scholar] [CrossRef] [PubMed]
- Klinger, J.R. Group III Pulmonary Hypertension: Pulmonary Hypertension Associated with Lung Disease: Epidemiology, Pathophysiology, and Treatments. Cardiol. Clin. 2016, 34, 413–433. [Google Scholar] [CrossRef]
- Garcia, A.R.; Torrubiano, I.V.; Blanco, I.; Estepar, R.S.J.; Chiaradia, D.A.R.; Ontiyuelo, C.M.; Meseguer, M.L.; Nardelli, P.; Gonzales, F.H.; Ribas, J.; et al. Reduced Pulmonary Vascular Density in Severe Pulmonary Hypertension Associated with Chronic Lung Disease. Eur. Respir. J. 2023, 62 (Suppl. S67), PA3491. [Google Scholar] [CrossRef]
- Piccari, L.; Blanco, I.; Torralba, Y.; Arismendi, E.; Gistau, C.; Ramírez, A.; Gimeno-Santos, E.; Roca, J.; Burgos, F.; Rodríguez-Roisín, R.; et al. Mechanisms of hypoxaemia in severe pulmonary hypertension associated with COPD. Eur. Respir. J. 2023, 62, 2300463. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Received PAH-Specific Therapy (n = 123) | Did Not Receive PAH-Specific Therapy (n = 660) | |
|---|---|---|
| Age, years (SD) * | 61.2 (10.8) | 63.8 (9.7) |
| Gender | ||
| Male, n (%) | 57 (46.3) | 347 (52.6) |
| Female, n (%) | 66 (53.7) | 313 (47.4) |
| BMI, kg/m2 (SD) | 29.3 (7.9) | 29.0 (7.0) |
| Race | ||
| White, n (%) | 60 (48.8) | 348 (52.7) |
| Black, n (%) | 51 (41.5) | 227 (34.4) |
| Hispanic, n (%) | 10 (8.1) | 64 (9.7) |
| Other, n (%) | 2 (1.6) | 21 (3.2) |
| Underlying lung disease * | ||
| COPD, n (%) | 35 (28.4) | 294 (44.5) |
| IPF, n (%) | 24 (19.5) | 104 (15.8) |
| Other pulmonary fibrosis, n (%) | 23 (18.7) | 93 (14.1) |
| Non-fibrotic ILD, n (%) | 7 (5.7) | 29 (4.4) |
| Sarcoidosis, n (%) | 22 (17.9) | 48 (7.3) |
| CPFE, n (%) | 12 (9.8) | 92 (13.9) |
| Cardiac disease, n (%) | 77 (62.6) | 422 (63.9) |
| Chronic kidney disease, n (%) * | 64 (52.0) | 416 (63.0) |
| Diabetes, n (%) | 37 (30.0) | 224 (33.9) |
| Smoking history, n (%) | 32 (26.0) | 191 (28.9) |
| Received PAH-Specific Therapy (n = 123) | Did Not Receive PAH-Specific Therapy (n = 660) | |
|---|---|---|
| FEV1, L (SD) | 1.4 (0.6) | 1.3 (0.7) |
| FVC, L (SD) | 2.0 (0.8) | 2.1 (0.8) |
| Percent predicted DLCO (SD) | 27.6 (15.4) | 28.1 (15.2) |
| mRAP, mmHg (SD) | 7.9 (5.3) | 7.1 (4.6) |
| sPAP, mmHg (SD) * | 66.6 (19.5) | 45.4 (13.9) |
| mPAP, mmHg (SD) * | 41.1 (11.3) | 29.3 (8.7) |
| mPAWP, mmHg (SD) | 11.9 (5.6) | 12.6 (11.4) |
| CO, L/min (SD) | 4.7 (3.8) | 5.1 (2.4) |
| CI, L/min/m2 (SD) * | 2.3 (0.6) | 2.6 (0.7) |
| PVR, WU (SD) * | 8.3 (5.4) | 4.4 (4.3) |
| Severe PH, n (%) * | 88 (71.5) | 158 (23.9) |
| 6MWD, m (SD) | 228.7 (97.1) | 240.6 (95.7) |
| Oxygen requirements, L/min (SD) | 6.2 (5.5) | 5.5 (4.6) |
| BNP, pg/mL (SD) * | 711.3 (413.9) | 368.9 (276.7) |
| Received PAH-Specific Therapy (n = 123) | Did Not Receive PAH-Specific Therapy (n = 660) | |
|---|---|---|
| Death, n (%) | 28 (22.8) | 112 (17.0) |
| Lung transplantation, n (%) * | 47 (38.2) | 383 (58.0) |
| Alive at last follow-up without a lung transplant, n (%) * | 48 (39.0) | 165 (25.0) |
| Median survival time from PH diagnosis, months (IQR) | 98.5 (61.9, 135.2) | 96.2 (90.4, 102.1) |
| Received PAH-Specific Therapy (n = 88) | Did Not Receive PAH-Specific Therapy (n = 158) | |
|---|---|---|
| FEV1, L (SD) | 1.4 (0.6) | 1.4 (0.6) |
| FVC, L (SD) | 2.2 (0.9) | 2.1 (0.8) |
| DLCO < 40% predicted, n (%) | 80 (90.9) | 137 (86.7) |
| mRAP, mmHg (SD) | 7.2 (5.9) | 7.5 (4.9) |
| sPAP, mmHg (SD) | 62.1 (17.5) | 63.9 (17.9) |
| mPAP, mmHg (SD) | 39.1 (11.6) | 40.2 (10.3) |
| mPAWP, mmHg (SD) | 11.1 (7.4) | 11.3 (6.8) |
| CO, L/min (SD) | 4.6 (2.7) | 4.5 (2.7) |
| CI, L/min/m2 (SD) | 2.2 (0.6) | 2.3 (0.6) |
| PVR, WU (SD) | 11.3 (10.7) | 8.5 (4.2) |
| 6MWD, m (SD) | ||
| <150 m | 17 (19.3) | 40 (25.5) |
| 150–300 m | 54 (61.4) | 89 (56.2) |
| >300 m | 17 (19.3) | 29 (18.3) |
| Hypoxemic, n (%) | 77 (87.5) | 128 (81.0) |
| BNP > 100 pg/mL, n (%) * | 53 (60.2) | 120 (75.9) |
| Received PAH-Specific Therapy (n = 88) | Did Not Receive PAH-Specific Therapy (n = 158) | |
|---|---|---|
| Death, n (%) | 22 (25.0) | 49 (31.0) |
| Lung transplantation, n (%) * | 28 (31.8) | 79 (50.0) |
| Alive at the last follow-up without a lung transplant, n (%) * | 38 (43.2) | 30 (19.0) |
| Median survival time from PH diagnosis, months (IQR) * | 119.1 (68.0, 170.2) | 48.9 (39.7, 58.0) |
| Variable | Association with Mortality |
|---|---|
| PAH-specific therapy | HR 0.31, 95% CI 0.11–0.88, p = 0.03 * |
| Underlying lung disease (COPD as reference) | |
| IPF | HR 0.46, 95% CI 0.15–1.44, p = 0.18 |
| Other pulmonary fibrosis | HR 0.78, 95% CI 0.23–2.65, p = 0.70 |
| Non-fibrotic ILD | HR 1.72, 95% CI 0.19–15.31, p = 0.63 |
| Sarcoidosis | HR 0.38, 95% CI 0.10–1.40, p = 0.15 |
| CPFE | HR 0.93, 95% CI 0.30–2.86, p = 0.90 |
| FVC | HR 0.95, 95% CI 0.91–1.15, p = 0.68 |
| DLCO < 40% predicted | HR 1.25, 95% CI 1.11–1.45, p = 0.048 * |
| 6MWD (compared to walking distance > 300 m) | |
| <150 m | HR 1.44, 95% CI 0.47–4.41, p = 0.52 |
| 150–300 m | HR 1.38, 95% CI 0.50–3.80, p = 0.53 |
| Hypoxemia | HR 2.23, 95% CI 0.86–5.75, p = 0.10 |
| BNP > 100 pg/mL | HR 0.92, 95% CI 0.41–2.09, p = 0.85 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schanz, O.; Criner, G.J.; Rali, P.; Gayen, S. Pulmonary Vasodilator Therapy Is Associated with Decreased Mortality in Patients with Chronic Lung Disease and Severe Pulmonary Hypertension. J. Cardiovasc. Dev. Dis. 2024, 11, 89. https://doi.org/10.3390/jcdd11030089
Schanz O, Criner GJ, Rali P, Gayen S. Pulmonary Vasodilator Therapy Is Associated with Decreased Mortality in Patients with Chronic Lung Disease and Severe Pulmonary Hypertension. Journal of Cardiovascular Development and Disease. 2024; 11(3):89. https://doi.org/10.3390/jcdd11030089
Chicago/Turabian StyleSchanz, Olivia, Gerard J. Criner, Parth Rali, and Shameek Gayen. 2024. "Pulmonary Vasodilator Therapy Is Associated with Decreased Mortality in Patients with Chronic Lung Disease and Severe Pulmonary Hypertension" Journal of Cardiovascular Development and Disease 11, no. 3: 89. https://doi.org/10.3390/jcdd11030089
APA StyleSchanz, O., Criner, G. J., Rali, P., & Gayen, S. (2024). Pulmonary Vasodilator Therapy Is Associated with Decreased Mortality in Patients with Chronic Lung Disease and Severe Pulmonary Hypertension. Journal of Cardiovascular Development and Disease, 11(3), 89. https://doi.org/10.3390/jcdd11030089