Hemodynamics in Cardiac Development


Abstract
:1. Introduction
2. Cardiac Anomalies after Vitelline Vein Ligation
Materials and Methods
3. Results
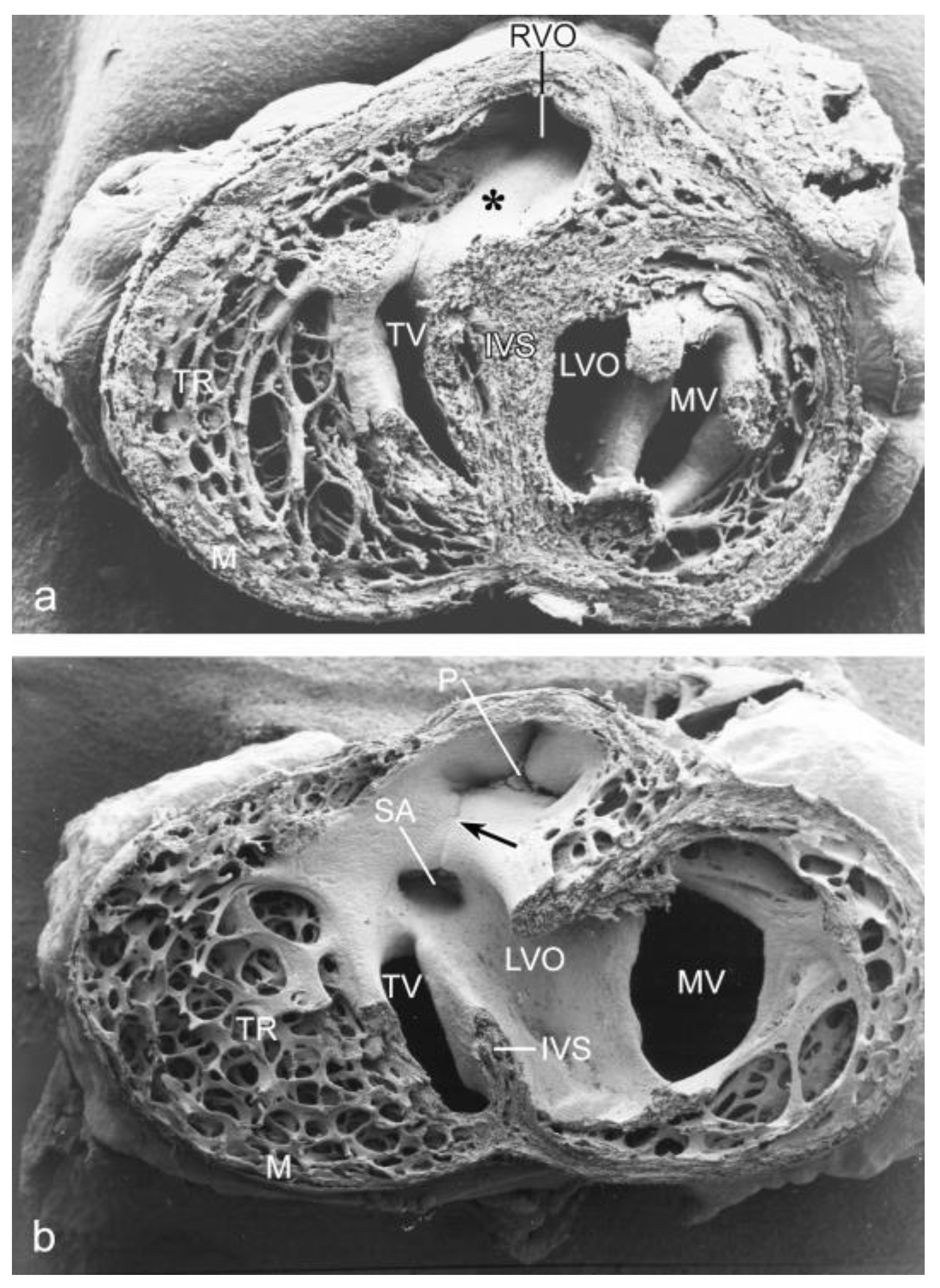
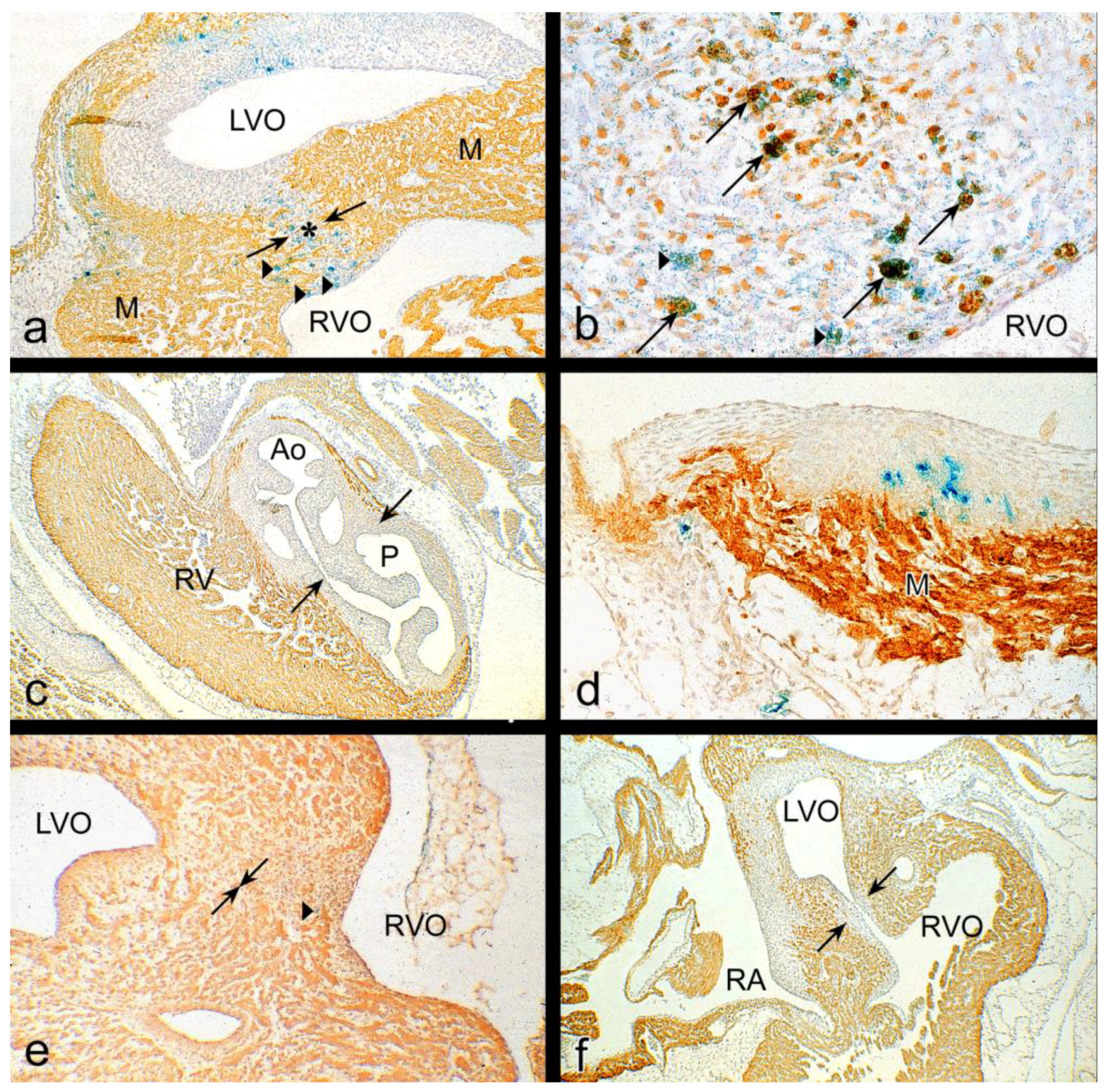
3.1. Impaired Development in Preseptation Stages and Tgfβ Receptor III (TBRIII) Expression
3.2. Peri- and Post-Septation Stages
4. Discussion
4.1. Hemodynamic Load
4.2. Ciliary Mechanosensing
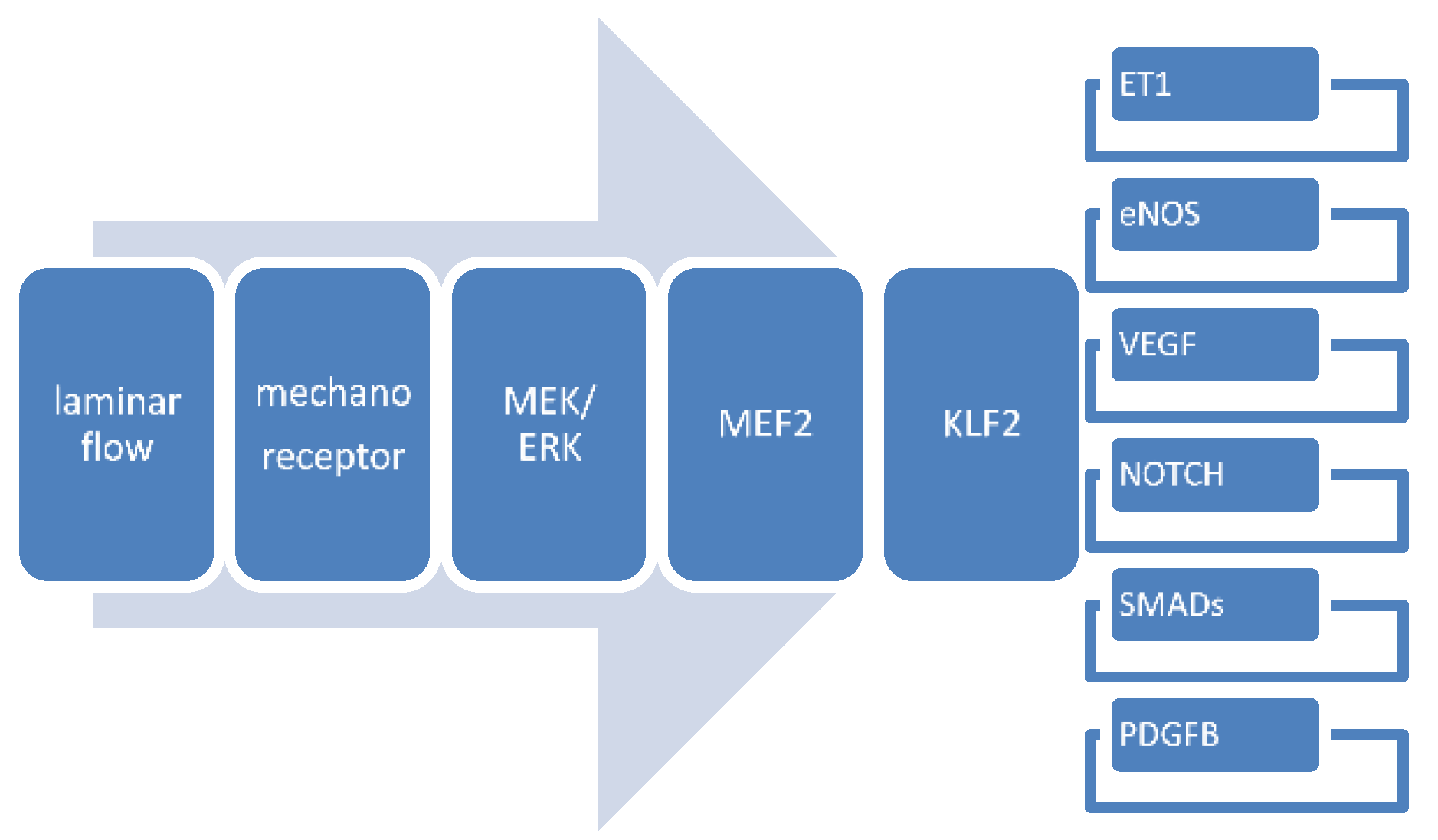
4.3. Gene Expression Patterns in Mechanosensing
4.3.1. Vascular Endothelial Growth Factor (Vegf) Signaling
4.3.2. Notch Signaling
4.3.3. Platelet-Derived Growth Factor (Pdgf) Signaling
4.3.4. Krüppel-Like Factor-2
4.3.5. Endothelin Signaling
4.3.6. Nitric Oxide (NO) Signaling
4.3.7. Tgfβ/Bmp Signaling
4.3.8. Interactions between Flow-Responsive Genes
4.4. Consequences for Human Embryonic Development
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gittenberger-de Groot, A.C.; Calkoen, E.E.; Poelmann, R.E.; Bartelings, M.M.; Jongbloed, M.R. Morphogenesis and molecular considerations on congenital cardiac septal defects. Ann. Med. 2014, 46, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Hogers, B.; DeRuiter, M.C.; Gittenberger-de Groot, A.C.; Poelmann, R.E. Unilateral vitelline vein ligation alters intracardiac blood flow patterns and morphogenesis in the chick embryo. Circ. Res. 1997, 80, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Groenendijk, B.C.; Hierck, B.P.; Vrolijk, J.; Baiker, M.; Pourquie, M.J.; Gittenberger-de Groot, A.C.; Poelmann, R.E. Changes in shear stress-related gene expression after experimentally altered venous return in the chicken embryo. Circ. Res. 2005, 96, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.G. The second heart field. Curr. Top. Dev. Biol. 2012, 100, 33–65. [Google Scholar] [CrossRef] [PubMed]
- Groenendijk, B.C.; Van der Heiden, K.; Hierck, B.P.; Poelmann, R.E. The role of shear stress on ET-1, KLF2, and NOS-3 expression in the developing cardiovascular system of chicken embryos in a venous ligation model. Physiology 2007, 22, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Okagawa, H.; Markwald, R.R.; Sugi, Y. Functional BMP receptor in endocardial cells is required in atrioventricular cushion mesenchymal cell formation in chick. Dev. Biol. 2007, 306, 179–928. [Google Scholar] [CrossRef] [PubMed]
- Gittenberger-de Groot, A.C.; Vrancken Peeters, M.P.; Mentink, M.M.; Gourdie, R.G.; Poelmann, R.E. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ. Res. 1998, 82, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pomares, J.M.; Phelps, A.; Sedmerova, M.; Carmona, R.; González-Iriarte, M.; Muñoz-Chápuli, R.; Wessels, A. Experimental studies on the spatiotemporal expression of WT1 and RALDH2 in the embryonic avian heart: A model for the regulation of myocardial and valvuloseptal development by epicardially derived cells (EPDCs). Dev. Biol. 2002, 247, 307–326. [Google Scholar] [CrossRef] [PubMed]
- Poelmann, R.E.; Mikawa, T.; Gittenberger-de Groot, A.C. Neural crest cells in outflow tract septation of the embryonic chicken heart: Differentiation and apoptosis. Dev. Dyn. 1998, 212, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Waldo, K.; Miyagawa-Tomita, S.; Kumiski, D.; Kirby, M.L. Cardiac neural crest cells provide new insight into septation of the cardiac outflow tract: Aortic sac to ventricular septal closure. Dev. Biol. 1998, 196, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Goddeeris, M.M.; Schwartz, R.; Klingensmith, J.; Meyers, E.N. Independent requirements for Hedgehog signaling by both the anterior heart field and neural crest cells for outflow tract development. Development 2007, 134, 1593–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, J.C.; Chughtai, M.; Wisse, L.J.; Gittenberger-de Groot, A.C.; Feng, Q.; Goumans, M.T.H.; VanMunsteren, J.C.; Jongbloed, M.R.M.; DeRuiter, M.C. Nos3 mutation leads to abnormal neural crest cell and second heart field lineage patterning in bicuspid aortic valve formation. Dis. Model. Mech. 2018, Dmm.034637. [Google Scholar] [CrossRef]
- Gong, H.; Lyu, X.; Wang, Q.; Hu, M.; Zhang, X. Endothelial to mesenchymal transition in the cardiovascular system. Life Sci. 2017, 184, 95–102. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Agbu, S.; Anderson, K.V. Microtubule Motors Drive Hedgehog Signaling in Primary Cilia. Trends Cell Biol. 2017, 27, 110–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, J.A.; Young, K.E.; Beachy, P.A. Cholesterol modification of hedgehog signaling proteins in animal development. Science 1996, 274, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Hildreth, V.; Webb, S.; Chaudhry, B.; Peat, J.D.; Phillips, H.M.; Brown, N.; Anderson, R.H.; Henderson, D.J. Left cardiac isomerism in the Sonic hedgehog null mouse. J. Anat. 2009, 214, 894–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yashiro, K.; Shiratori, H.; Hamada, H. Haemodynamics determined by a genetic programme govern asymmetric development of the aortic arch. Nature 2007, 450, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Hogers, B.; DeRuiter, M.C.; Baasten, A.M.; Gittenberger-de Groot, A.C.; Poelmann, R.E. Intracardiac blood flow patterns related to the yolk sac circulation of the chick embryo. Circ. Res. 1995, 76, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Rugonyi, S.; Shaut, C.; Liu, A.; Thornburg, K.; Wang, R. Changes in wall motion and blood flow in the outflow tract of chick embryonic hearts observed with optical coherence tomography after outflow tract banding and vitelline-vein ligation. Phys. Med. Biol. 2008, 53, 5077–5091. [Google Scholar] [CrossRef] [PubMed]
- Poelma, C.; Van der Heiden, K.; Hierck, B.P.; Poelmann, R.E.; Westerweel, J. Measurements of the wall shear stress distribution in the outflow tract of an embryonic chicken heart. J. R. Soc. Interface 2010, 7, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalski, W.J.; Teslovich, N.C.; Menon, P.G.; Tinney, J.P.; Keller, B.B.; Pekkan, K. Left atrial ligation alters intracardiac flow patterns and the biomechanical landscape in the chick embryo. Dev. Dyn. 2014, 243, 652–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamison, R.A.; Samarage, C.R.; Bryson-Richardson, R.J.; Fouras, A. In vivo wall shear measurements within the developing zebrafish heart. PLoS ONE 2013, 8, e75722. [Google Scholar] [CrossRef] [PubMed]
- Kalogirou, S.; Malissovas, N.; Moro, E.; Argenton, F.; Stainier, D.Y.; Beis, D. Intracardiac flow dynamics regulate atrioventricular valve morphogenesis. Cardiovasc. Res. 2014, 104, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, T.A.; Robinson, J.Y.; How, T.; DeLaughter, D.M.; Blobe, G.C.; Barnett, J.V. Endocardial cell epithelial-mesenchymal transformation requires Type III TGFβ receptor interaction with GIPC. Cell Signal. 2012, 24, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egorova, A.D.; Van der Heiden, K.; Van de Pas, S.; Vennemann, P.; Poelma, C.; DeRuiter, M.C.; Goumans, M.J.; Gittenberger-de Groot, A.C.; Ten Dijke, P.; Poelmann, R.E.; et al. Tgfβ/Alk5 signaling is required for shear stress induced klf2 expression in embryonic endothelial cells. Dev. Dyn. 2011, 240, 1670–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milgrom-Hoffman, M.; Michailovici, I.; Ferrara, N.; Zelzer, E.; Tzahor, E. Endothelial cells regulate neural crest and second heart field morphogenesis. Biol. Open 2014, 3, 679–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.J.; Watanabe, Y.; Smyth, G.; Miyagawa-Tomita, S.; Meyers, E.; Klingensmith, J.; Camenisch, T.; Buckingham, M.; Moon, A.M. An FGF autocrine loop initiated in second heart field mesoderm regulates morphogenesis at the arterial pole of the heart. Development 2008, 135, 3599–3610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piek, E.; Heldin, C.H.; Ten Dijke, P. Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 1999, 13, 2105–2124. [Google Scholar] [CrossRef] [PubMed]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [PubMed]
- Bartram, U.; Molin, D.G.; Wisse, L.J.; Mohamad, A.; Sanford, L.P.; Doetschman, T.; Speer, C.P.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-beta(2)-knockout mice. Circulation 2001, 103, 2745–2752. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.B.; Boyer, A.S.; Runyan, R.B.; Barnett, J.V. Requirement of type III TGF-beta receptor for endocardial cell transformation in the heart. Science 1999, 283, 2080–2082. [Google Scholar] [CrossRef] [PubMed]
- Broekhuizen, M.L.; Hogers, B.; DeRuiter, M.C.; Poelmann, R.E.; Gittenberger-de Groot, A.C.; Wladimiroff, J.W. Altered hemodynamics in chick embryos after extraembryonic venous obstruction. Ultrasound Obstet. Gynecol. 1999, 13, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.H.; Chaudhry, B.; Mohun, T.J.; Bamforth, S.D.; Hoyland, D.; Phillips, H.M.; Webb, S.; Moorman, A.F.; Brown, N.A.; Henderson, D.J. Normal and abnormal development of the intrapericardial arterial trunks in humans and mice. Cardiovasc. Res. 2012, 95, 108–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar] [PubMed]
- Bartelings, M.M.; Gittenberger-de Groot, A.C. The outflow tract of the heart-embryologic and morphologic correlations. Int. J. Cardiol. 1989, 22, 289–300. [Google Scholar] [CrossRef]
- Poelmann, R.E.; Molin, D.; Wisse, L.J.; Gittenberger-de Groot, A.C. Apoptosis in cardiac development. Cell Tissue Res. 2000, 301, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Brauer, P.R.; Yee, J.A. Cranial neural crest cells synthesize and secrete a latent form of transforming growth factor beta that can be activated by neural crest cell proteolysis. Dev. Biol. 1993, 155, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Brauer, P.R. Latent transforming growth factor-beta is present in the extracellular matrix of embryonic hearts in situ. Dev. Dyn. 1996, 205, 126–134. [Google Scholar] [CrossRef]
- Ramsdell, A.F.; Markwald, R.R. Induction of endocardial cushion tissue in the avian heart is regulated, in part, by TGFbeta-3-mediated autocrine signaling. Dev. Biol. 1997, 188, 64–74. [Google Scholar] [CrossRef] [PubMed]
- MacLellan, W.R.; Brand, T.; Schneider, M.D. Transforming growth factor-beta in cardiac ontogeny and adaptation. Circ. Res. 1993, 73, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Takamura, K.; Okishima, T.; Ohdo, S.; Hayakawa, K. Association of cephalic neural crest cells with cardiovascular development, particularly that of the semilunar valves. Anat. Embryol. 1990, 182, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, H.C.; Shekhar, A.; McQuinn, T.C.; Butcher, J.T. Hemodynamic patterning of the avian atrioventricular valve. Dev. Dyn. 2011, 240, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, S.E.; Butcher, J.T.; Yalcin, H.C. Mechanical regulation of cardiac development. Front. Physiol. 2014, 5, 318. [Google Scholar] [CrossRef] [PubMed]
- Midgett, M.; Rugonyi, S. Congenital heart malformations induced by hemodynamic altering surgical interventions. Front. Physiol. 2014, 5, 287. [Google Scholar] [CrossRef] [PubMed]
- Sedmera, D.; Pexieder, T.; Rychterova, V.; Hu, N.; Clark, E.B. Remodeling of chick embryonic ventricular myoarchitecture under experimentally changed loading conditions. Anat. Rec. 1999, 254, 238–252. [Google Scholar] [CrossRef] [Green Version]
- Pesevski, Z.; Kvasilova, A.; Stopkova, T.; Nanka, O.; Drobna Krejci, E.; Buffinton, C.; Kockova, R.; Eckhardt, A.; Sedmera, D. Endocardial Fibroelastosis is Secondary to Hemodynamic Alterations in the Chick Embryonic Model of Hypoplastic Left Heart Syndrome. Dev. Dyn. 2018, 247, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Christensen, D.A.; Agrawal, A.K.; Beaumont, C.; Clark, E.B.; Hawkins, J.A. Dependence of aortic arch morphogenesis on intracardiac blood flow in the left atrial ligated chick embryo. Anat. Rec. 2009, 292, 652–660. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, A.; McQuinn, T.; Sedmera, D. Increased ventricular preload is compensated by myocyte proliferation in normal and hypoplastic fetal chick left ventricle. Circ. Res. 2007, 100, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Tobita, K.; Garrison, J.B.; Liu, L.J.; Tinney, J.P.; Keller, B.B. Three-dimensional myofiber architecture of the embryonic left ventricle during normal development and altered mechanical loads. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2005, 283, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midgett, M.; López, C.S.; David, L.; Maloyan, A.; Rugonyi, S. Increased Hemodynamic Load in Early Embryonic Stages Alters Endocardial to Mesenchymal Transition. Front. Physiol. 2017, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Ford, S.M.; McPheeters, M.T.; Wang, Y.T.; Ma, P.; Gu, S.; Strainic, J.; Snyder, C.; Rollins, A.M.; Watanabe, M.; Jenkins, M.W. Increased regurgitant flow causes endocardial cushion defects in an avian embryonic model of congenital heart disease. Congenit. Heart Dis. 2017, 12, 322–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alenghat, F.J.; Ingber, D.E. Mechanotransduction: All signals point to cytoskeleton, matrix, and integrins. Sci. STKE 2002, 119, pe6. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalo, F.R.; Corbit, K.C.; Sirerol-Piquer, M.S.; Ramaswami, G.; Otto, E.A.; Noriega, T.R.; Seol, A.D.; Robinson, J.F.; Bennett, C.L.; Josifova, D.J.; et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet. 2011, 43, 7767–7784. [Google Scholar] [CrossRef] [PubMed]
- Eggenschwiler, J.T.; Anderson, K.V. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 2007, 23, 345–373. [Google Scholar] [CrossRef] [PubMed]
- Malicki, J.J.; Johnson, C.A. The Cilium: Cellular Antenna and Central Processing Unit. Trends Cell Biol. 2017, 27, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Emmer, B.T.; Maric, D.; Engman, D.M. Molecular mechanisms of protein and lipid targeting to ciliary membranes. J. Cell Sci. 2010, 123, 529–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maimari, N.; Pedrigi, R.M.; Russo, A.; Broda, K.; Krams, R. Integration of flow studies for robust selection of mechanoresponsive genes. Thromb. Haemost. 2016, 115, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Slough, J.; Cooney, L.; Brueckner, M. Monocilia in the embryonic mouse heart suggest a direct role for cilia in cardiac morphogenesis. Dev. Dyn. 2008, 237, 2304–2314. [Google Scholar] [CrossRef] [PubMed]
- Hierck, B.P.; Van der Heiden, K.; Alkemade, F.E.; Van de Pas, S.; Van Thienen, J.V.; Groenendijk, B.C.; Bax, W.H.; Van der Laarse, A.; DeRuiter, M.C.; Horrevoets, A.J.; et al. Primary cilia sensitize endothelial cells for fluid shear stress. Dev. Dyn. 2008, 237, 725–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Heiden, K.; Egorova, A.D.; Poelmann, R.E.; Wentzel, J.J.; Hierck, B.P. Role for primary cilia as flow detectors in the cardiovascular system. Int. Rev. Cell Mol. Biol. 2011, 290, 87–119. [Google Scholar] [CrossRef] [PubMed]
- Van der Heiden, K.; Hierck, B.P.; Krams, R.; de Crom, R.; Cheng, C.; Baiker, M.; Pourquie, M.J.; Alkemade, F.E.; DeRuiter, M.C.; Gittenberger-de Groot, A.C.; et al. Endothelial primary cilia in areas of disturbed flow are at the base of atherosclerosis. Atherosclerosis 2008, 196, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Egorova, A.D.; Khedoe, P.P.; Goumans, M.J.; Yoder, B.K.; Nauli, S.M.; Ten Dijke, P.; Poelmann, R.E.; Hierck, B.P. Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition. Circ. Res. 2011, 108, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Hogers, B.; DeRuiter, M.C.; Gittenberger-de Groot, A.C.; Poelmann, R.E. Extraembryonic venous obstructions lead to cardiovascular malformations and can be embryolethal. Cardiovasc. Res. 1999, 41, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Collen, D. Molecular basis of angiogenesis. Role of VEGF and VE-cadherin. Ann. N. Y. Acad. Sci. 2000, 902, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Van den Akker, N.M.; Caolo, V.; Molin, D.G. Cellular decisions in cardiac outflow tract and coronary development: An act by VEGF and NOTCH. Differentiation 2012, 84, 62–78. [Google Scholar] [CrossRef] [PubMed]
- Dor, Y.; Camenisch, T.D.; Itin, A.; Fishman, G.I.; McDonald, J.A.; Carmeliet, P.; Keshet, E. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development 2001, 128, 1531–1538. [Google Scholar] [PubMed]
- Smedts, H.P.; Isaacs, A.; de Costa, D.; Uitterlinden, A.G.; van Duijn, C.M.; Gittenberger-de Groot, A.C.; Helbing, W.A.; Steegers, E.A.; Steegers-Theunissen, P. VEGF polymorphisms are associated with endocardial cushion defects: A family-based case-control study. Pediatr. Res. 2010, 67, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Van den Akker, N.M.; Molin, D.G.; Peters, P.P.; Maas, S.; Wisse, L.J.; Van Brempt, R.; Van Munsteren, C.J.; Bartelings, M.M.; Poelmann, R.E.; Carmeliet, P.; et al. Tetralogy of Fallot and alterations in vascular endothelial growth factor-A signaling and notch signaling in mouse embryos solely expressing the VEGF120 isoform. Circ. Res. 2007, 100, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Jahnsen, E.D.; Trindade, A.; Zaun, H.C.; Lehoux, S.; Duarte, A.; Jones, E.A. Notch1 is pan-endothelial at the onset of flow and regulated by flow. PLoS ONE 2015, 10, e0122622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mack, J.J.; Iruela-Arispe, M.L. NOTCH regulation of the endothelial cell phenotype. Curr. Opin. Hematol. 2018, 25, 212–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amato, G.; Luxán, G.; del Monte-Nieto, G.; Martínez-Poveda, B.; Torroja, C.; Walter, W.; Bochter, M.S.; Benedito, R.; Cole, S.; Martinez, F.; et al. Sequential Notch activation regulates ventricular chamber development. Nat. Cell Biol. 2016, 18, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Iso, T.; Hamamori, Y.; Kedes, L. Notch signaling in vascular development. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Engleka, K.A.; Rentschler, S.L.; Manderfield, L.J.; Li, L.; Yuan, L.; Epstein, J.A. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J. Clin. Investig. 2011, 121, 422–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, R.C.; Laney, A.O.; Smith, R.; Gerfen, J.; Morrissette, J.J.; Woyciechowski, S.; Garbarini, J.; Loomes, K.M.; Krantz, I.D.; Urban, Z.; et al. Jagged1 (JAG1) mutations in patients with tetralogy of Fallot or pulmonic stenosis. Hum. Mutat. 2010, 31, 594–601. [Google Scholar] [CrossRef] [PubMed]
- McCright, B.; Lozier, J.; Gridley, T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development 2002, 129, 1075–1082. [Google Scholar] [PubMed]
- Schneider, L.; Clement, C.A.; Teilmann, S.C.; Pazour, G.J.; Hoffmann, E.K.; Satir, P.; Christensen, S.T. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 2005, 15, 1861–1866. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, R.; Gaetano, C.; Antonini, A.; Pompilio, G.; Bracco, E.; Rönnstrand, L.; Heldin, C.H.; Capogrossi, M.C. Different effects of high and low shear stress on platelet-derived growth factor isoform release by endothelial cells: Consequences for smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Bleyl, S.B.; Saijoh, Y.; Bax, N.A.; Gittenberger-de Groot, A.C.; Wisse, L.J.; Chapman, S.C.; Hunter, J.; Shiratori, H.; Hamada, H.; Yamada, S.; et al. Dysregulation of the PDGFRA gene causes inflow tract anomalies including TAPVR: Integrating evidence from human genetics and model organisms. Hum. Mol. Genet. 2010, 19, 1286–1301. [Google Scholar] [CrossRef] [PubMed]
- Bax, N.A.; Lie-Venema, H.; Vicente-Steijn, R.; Bleyl, S.B.; Van Den Akker, N.M.; Maas, S.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Platelet-derived growth factor is involved in the differentiation of second heart field-derived cardiac structures in chicken embryos. Dev. Dyn. 2009, 238, 2658–2669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, R.J.; Van Soest, S.; Fontijn, R.D.; Salamanca, S.; De Groot, P.G.; VanBavel, E.; Pannekoek, H.; Horrevoets, A.J. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Krüppel-like factor (KLF2). Blood 2002, 100, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Nayak, L.; Lin, Z.; Jain, M.K. “Go with the flow”: How Krüppel-like factor 2 regulates the vasoprotective effects of shear stress. Antioxid. Redox Signal. 2011, 15, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Leyen, T.A.; Fontijn, R.D.; Fledderus, J.O.; Baggen, J.M.; Volger, O.L.; van Nieuw Amerongen, G.P.; Horrevoets, A.J. KLF2-induced actin shear fibers control both alignment to flow and JNK signaling in vascular endothelium. Blood 2010, 115, 2533–2542. [Google Scholar] [CrossRef] [PubMed]
- Fontijn, R.D.; Volger, O.L.; Van der Pouw-Kraan, T.C.; Doddaballapur, A.; Leyen, T.; Baggen, J.M.; Boon, R.; Horrevoets, A.J. Expression of Nitric Oxide-Transporting Aquaporin-1 Is Controlled by KLF2 and Marks Non-Activated Endothelium In Vivo. PLoS ONE 2015, 10, e0145777. [Google Scholar] [CrossRef] [PubMed]
- Chiplunkar, A.R.; Lung, T.K.; Alhashem, Y.; Koppenhaver, B.A.; Salloum, F.N.; Kukreja, R.C.; Haar, J.L.; Lloyd, J.A. Krüppel-like factor 2 is required for normal mouse cardiac development. PLoS ONE, 2013, 8, e54891. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Yu, Q.; Shin, J.T.; Sebzda, E.; Bertozzi, C.; Chen, M.; Mericko, P.; Stadtfeld, M.; Zhou, D.; Cheng, L.; et al. Klf2 is an essential regulator of vascular hemodynamic forces in vivo. Dev. Cell 2006, 11, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.L.; Parnall, M.; Loughna, S. Effect of altered haemodynamics on the developing mitral valve in chick embryonic heart. J. Mol. Cell. Cardiol. 2017, 108, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Vozzi, F.; Bianchi, F.; Ahluwalia, A.; Domenici, C. Hydrostatic pressure and shear stress affect endothelin-1 and nitric oxide release by endothelial cells in bioreactors. Biotechnol. J. 2014, 9, 146–154. [Google Scholar] [CrossRef] [PubMed]
- White, S.J.; Hayes, E.M.; Lehoux, S.; Jeremy, J.Y.; Horrevoets, A.J.; Newby, A.C. Characterization of the differential response of endothelial cells exposed to normal and elevated laminar shear stress. J. Cell. Physiol. 2011, 226, 2841–2848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.X.; Cai, S.X.; Wang, P.Q.; Ouyang, K.Q.; Wang, Y.L.; Xu, S.R. Shear-induced changes in endothelin-1 secretion of microvascular endothelial cells. Microvasc. Res. 2002, 63, 209–217. [Google Scholar] [CrossRef]
- Groenendijk, B.C.; Stekelenburg-de Vos, S.; Vennemann, P.; Wladimiroff, J.W.; Nieuwstadt, F.T.; Lindken, R.; Westerweel, J.; Hierck, B.P.; Ursem, N.T.; Poelmann, R.E. The endothelin-1 pathway and the development of cardiovascular defects in the haemodynamically challenged chicken embryo. J. Vasc. Res. 2008, 45, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, Y.; Kurihara, H.; Oda, H.; Maemura, K.; Nagai, R.; Ishikawa, T.; Yazaki, Y. Aortic arch malformations and ventricular septal defect in mice deficient in endothelin-1. J. Clin. Investig. 1995, 96, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, T.; Silacci, P.; Harrison, V.J.; Hayoz, D. Nitric oxide synthase expression in endothelial cells exposed to mechanical forces. Hypertension 1998, 32, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Balligand, J.L.; Feron, O.; Dessy, C. eNOS activation by physical forces: From short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol. Rev. 2009, 89, 481–534. [Google Scholar] [CrossRef] [PubMed]
- Rudic, R.D.; Shesely, E.G.; Maeda, N.; Smithies, O.; Segal, S.S.; Sessa, W.C. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J. Clin. Investig. 1998, 101, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Song, W.; Lu, X.; Hamilton, J.A.; Lei, M.; Peng, T.; Yee, S.P. Development of heart failure and congenital septal defects in mice lacking endothelial nitric oxide synthase. Circulation 2002, 106, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lu, X.; Xiang, F.L.; Poelmann, R.E.; Gittenberger-de Groot, A.C.; Robbins, J.; Feng, Q. Nitric oxide synthase-3 deficiency results in hypoplastic coronary arteries and postnatal myocardial infarction. Eur. Heart J. 2014, 35, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Gittenberger-de Groot, A.C.; Winter, E.M.; Bartelings, M.M.; Goumans, M.J.; DeRuiter, M.C.; Poelmann, R.E. The arterial and cardiac epicardium in development, disease and repair. Differentiation 2012, 84, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Cooley, J.R.; Yatskievych, T.A.; Antin, P.B. Embryonic expression of the transforming growth factor beta ligand and receptor genes in chicken. Dev. Dyn. 2014, 243, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Azhar, M.; Schultz, J.; Grupp, I.; Dorn, G.W., 2nd; Meneton, P.; Molin, D.G.; Gittenberger-de Groot, A.C.; Doetschman, T. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev. 2003, 14, 391–407. [Google Scholar] [CrossRef] [Green Version]
- Doetschman, T.; Barnett, J.V.; Runyan, R.B.; Camenisch, T.D.; Heimar, R.L.; Granzier, H.L.; Conway, S.J.; Azhar, M. Transforming growth factor beta signaling in adult cardiovascular diseases and repair. Cell Tissue Res. 2012, 347, 203–223. [Google Scholar] [CrossRef] [PubMed]
- Kruithof, B.P.; Duim, S.N.; Moerkamp, A.T.; Goumans, M.J. TGFβ and BMP signaling in cardiac cushion formation: Lessons from mice and chicken. Differentiation 2012, 84, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Papoutsi, T.; Luna-Zurita, L.; Prados, B.; Zaffran, S.; De la Pompa, J.L. Bmp2 and Notch cooperate to pattern the embryonic endocardium. Development 2018, 145, dev163378. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, B.; Ito, Y.; Makita, T.; Sasaki, T.; Chai, Y.; Sucov, H.M. Cardiovascular malformations with normal smooth muscle differentiation in neural crest-specific type II TGFbeta receptor (Tgfbr2) mutant mice. Dev. Biol. 2006, 289, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Walshe, T.E.; De la Paz, N.G.; D’Amore, P.A. The role of shear-induced transforming growth factor-β signaling in the endothelium. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2608–2617. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Bohanan, C.S.; Neumann, J.C.; Lingrel, J.B. KLF2 transcription factor modulates blood vessel maturation through smooth muscle cell migration. J. Biol. Chem. 2008, 283, 3942–3950. [Google Scholar] [CrossRef] [PubMed]
- Dekker, R.J.; Boon, R.A.; Rondaij, M.G.; Kragt, A.; Volger, O.L.; Elderkamp, Y.W.; Meijers, J.C.; Voorberg, J.; Pannekoek, H.; Horrevoets, A.J. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood 2006, 107, 4354–4363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushton, D.I. Pathology of placenta. In Textbook of Fetal and Perinatal Pathology; Wigglesworth, J.S., Singer, D.B., Eds.; Blackwell Scientific Publications: Boston, MA, USA, 1991; pp. 161–220. [Google Scholar]
- Rosenthal, G.L. Patterns of prenatal growth among infants with cardiovascular malformations: Possible fetal hemodynamic effects. Am. J. Epidemiol. 1996, 143, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Groenenberg, I.A.; Wladimiroff, J.W.; Hop, W.C. Fetal cardiac and peripheral arterial flow velocity waveforms in intrauterine growth retardation. Circulation 1989, 80, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- Jantzen, D.W.; Moon-Grady, A.J.; Morris, S.A.; Armstrong, A.K.; Berg, C.; Dangel, J.; Fifer, C.G.; Frommelt, M.; Gembruch, U.; Herberg, U.; et al. Hypoplastic Left Heart Syndrome with Intact or Restrictive Atrial Septum: A Report From the International Fetal Cardiac Intervention Registry. Circulation 2017, 136, 1346–1349. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hamburger Hamilton (HH) Stages | 8–20 | 22–23 | 24 | Total 18–24 | ||||
|---|---|---|---|---|---|---|---|---|
| Number of Embryos | n = 19 | % | n = 16 | % | n = 28 | % | n = 61 | % |
| Normal | 0 | 0 | 0 | 0 | 77 | 25 | 7 | 11 |
| Disturbed looping | 6 | 32 | 5 | 31 | 9 | 32 | 20 | 32 |
| Hypoplastic atrioventricular (AV) cushions | 7 | 37 | 8 | 50 | 9 | 32 | 24 | 38 |
| Hypoplastic outflow tract (OFT) cushions | 3 | 16 | 2 | 13 | 6 | 21 | 11 | 17 |
| Myocardium left atrium | 8 | 42 | 4 | 25 | 0 | 0 | 16 | 25 |
| Myocardium ventricle | 3 | 16 | 3 | 19 | 6 | 21 | 12 | 19 |
| Pharyngeal arch arteries | 1 | 5 | 1 | 6 | 7 | 25 | 9 | 14 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poelmann, R.E.; Gittenberger-de Groot, A.C. Hemodynamics in Cardiac Development. J. Cardiovasc. Dev. Dis. 2018, 5, 54. https://doi.org/10.3390/jcdd5040054
Poelmann RE, Gittenberger-de Groot AC. Hemodynamics in Cardiac Development. Journal of Cardiovascular Development and Disease. 2018; 5(4):54. https://doi.org/10.3390/jcdd5040054
Chicago/Turabian StylePoelmann, Robert E., and Adriana C. Gittenberger-de Groot. 2018. "Hemodynamics in Cardiac Development" Journal of Cardiovascular Development and Disease 5, no. 4: 54. https://doi.org/10.3390/jcdd5040054
APA StylePoelmann, R. E., & Gittenberger-de Groot, A. C. (2018). Hemodynamics in Cardiac Development. Journal of Cardiovascular Development and Disease, 5(4), 54. https://doi.org/10.3390/jcdd5040054