Biomechanical Cues Direct Valvulogenesis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Structure and Function of Mature Valves
2. Valve Development
2.1. Linear Heart Tube Development
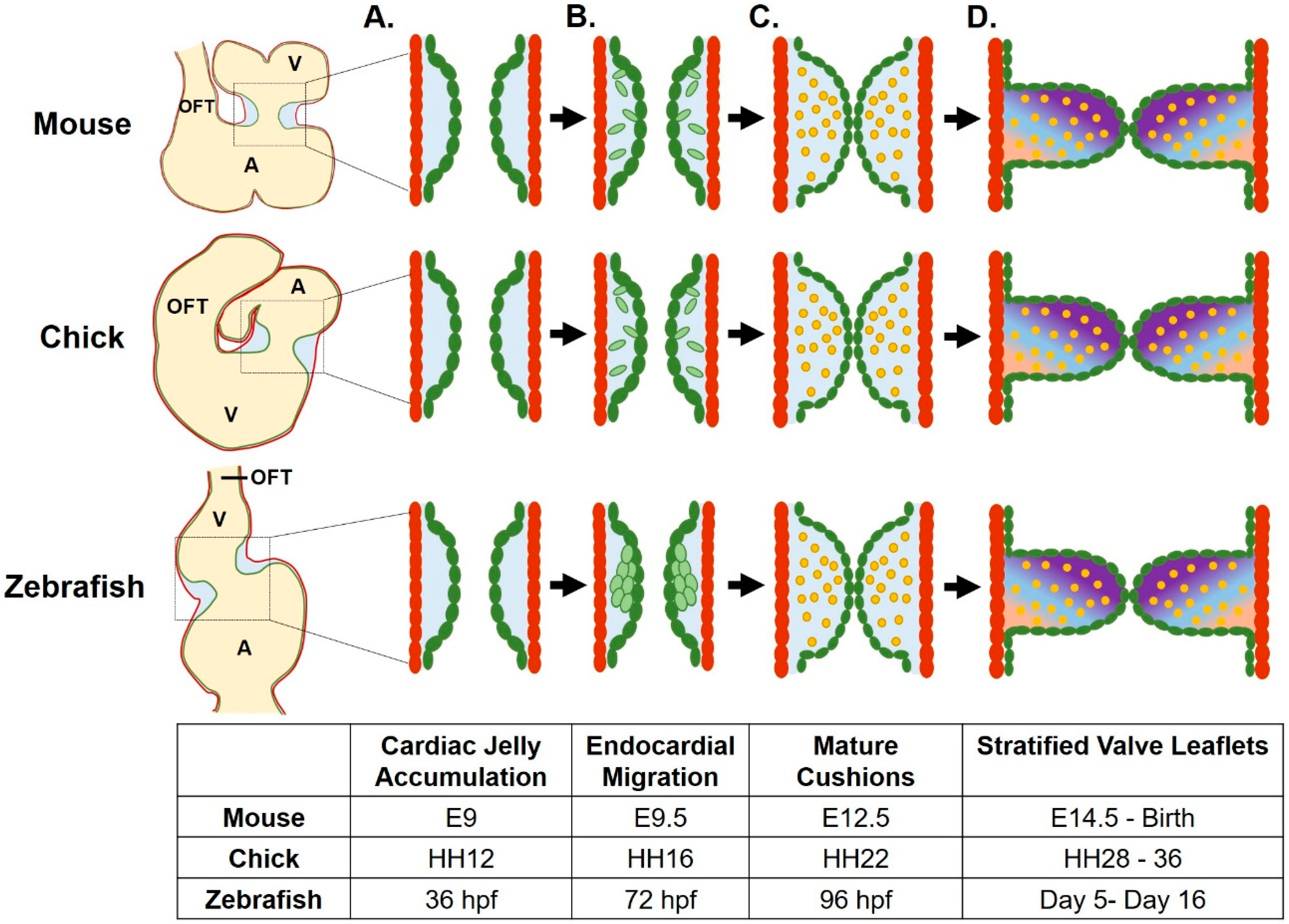
2.2. Endocardial Cushion Formation
2.3. Mesenchymal Cells Populate the Endocardial Cushions
2.4. Neural Crest Cells Contribute to the Development of the Semilunar Valves
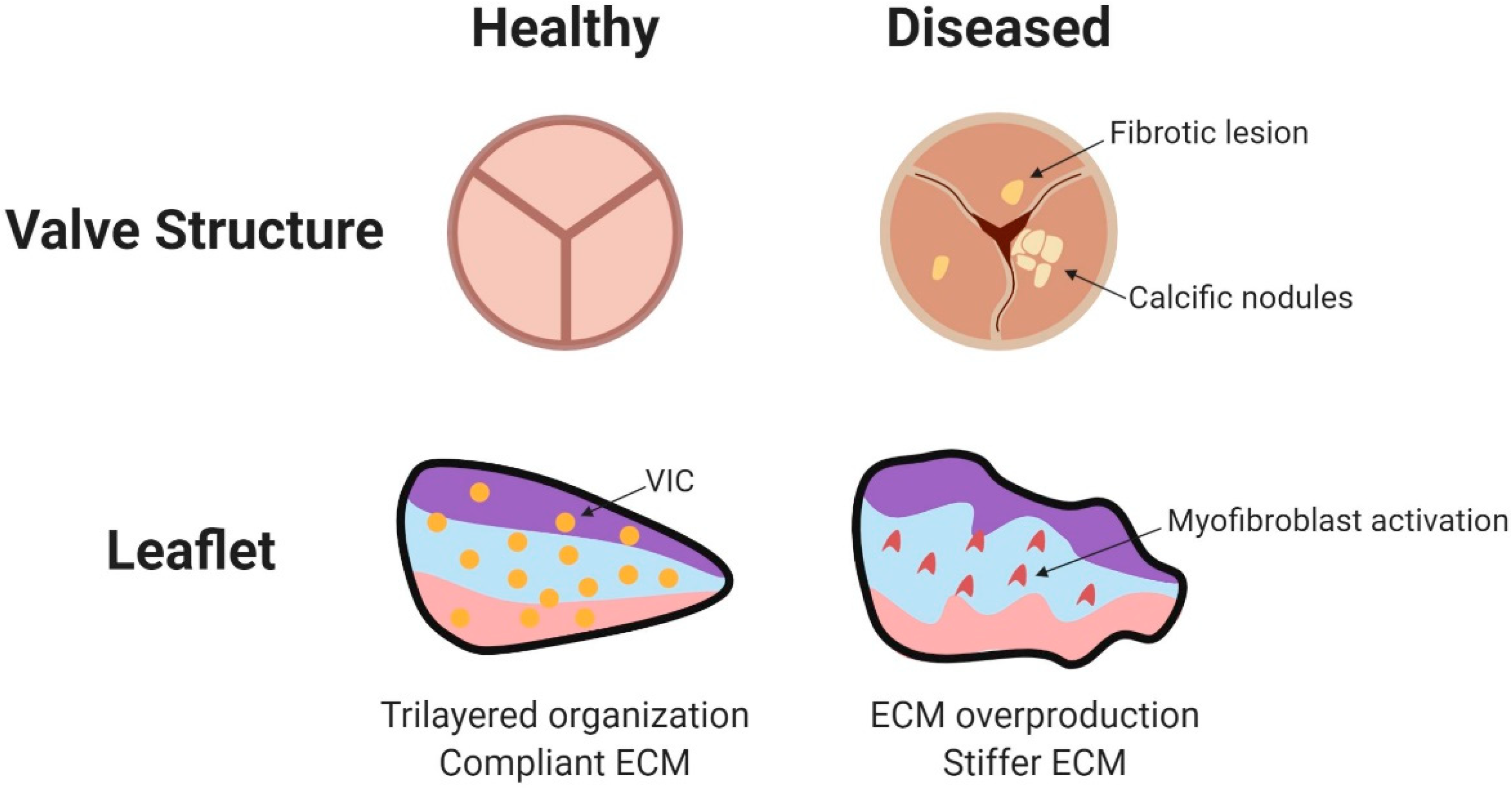
2.5. Valve Extracellular Matrix Remodeling
3. Biomechanical Inputs in Valve Development
3.1. Shear Stress
3.1.1. The KLF2 Pathway
3.1.2. The Role of Cilia in Sensing Shear Stress
3.1.3. MicroRNAs in the Shear Stress Response
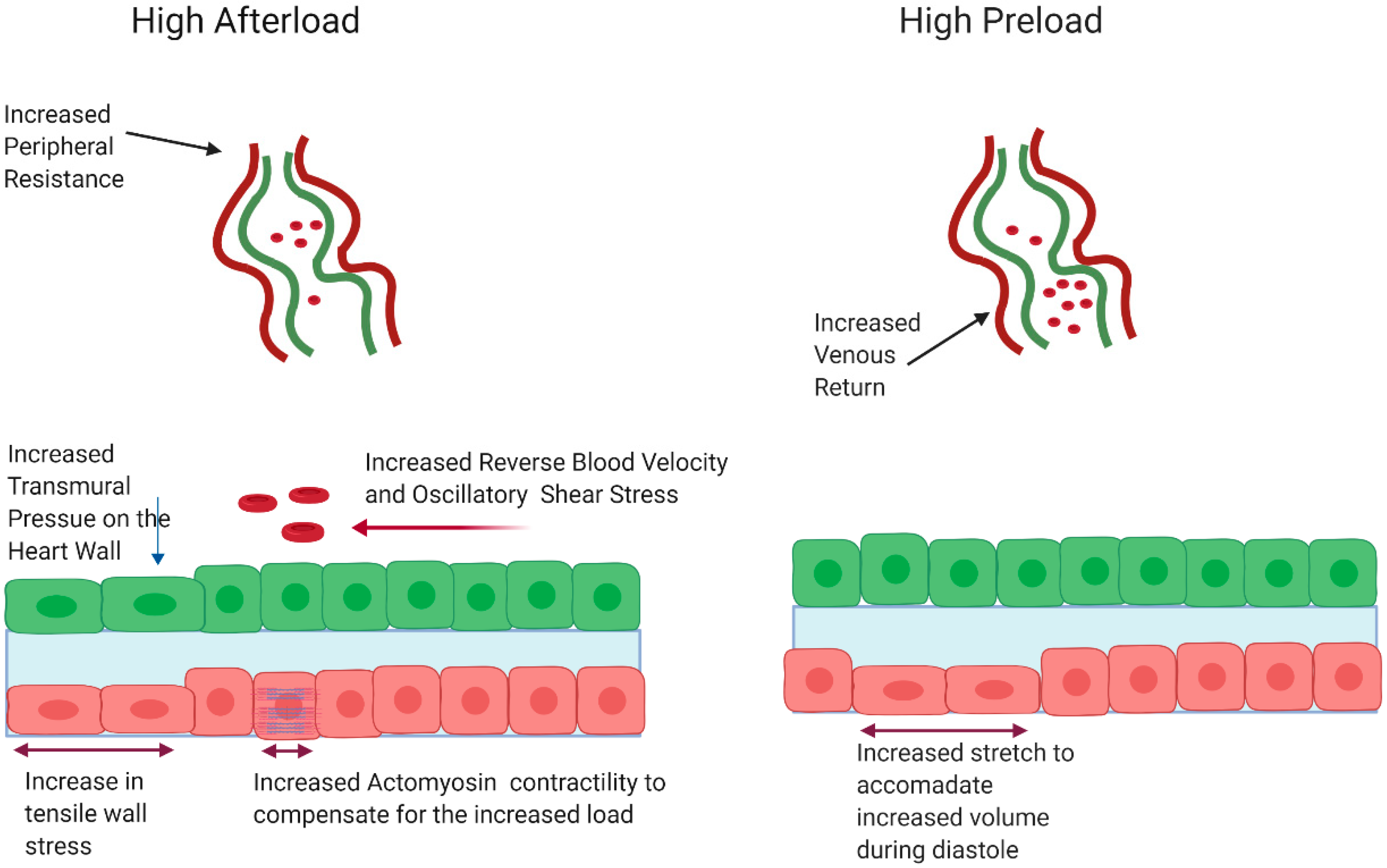
3.2. Pressure as a Hemodynamic Cue in Valve Development
3.2.1. Pressure in Adult Pathology
3.2.2. Pressure as a Cue during Embryonic Valve Development
3.2.3. Signaling Pathways Involved Sensing and Responding to Changes in Pressure in Valve Development
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Flanagan, T.C.; Pandit, A. Living artificial heart valve alternatives: A review. Eur. Cell Mater. 2003, 6, 28–45. [Google Scholar] [CrossRef]
- Schoen, F.J. Evolving concepts of cardiac valve dynamics: The continuum of development, functional structure, pathobiology, and tissue engineering. Circulation 2008, 118, 1864–1880. [Google Scholar] [CrossRef]
- Pozzoli, A.; Zuber, M.; Reisman, M.; Maisano, F.; Taramasso, M. Comparative Anatomy of Mitral and Tricuspid Valve: What Can the Interventionlist Learn From the Surgeon. Front. Cardiovasc. Med. 2018, 5, 80. [Google Scholar] [CrossRef]
- Lincoln, J.; Garg, V. Etiology of valvular heart disease-genetic and developmental origins. Circ. J. 2014, 78, 1801–1807. [Google Scholar] [CrossRef] [Green Version]
- Lincoln, J.; Alfieri, C.M.; Yutzey, K.E. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev. Dyn. 2004, 230, 239–250. [Google Scholar] [CrossRef]
- Dirkx, E.; da Costa Martins, P.A.; De Windt, L.J. Regulation of fetal gene expression in heart failure. Biochim. Biophys. Acta 2013, 1832, 2414–2424. [Google Scholar] [CrossRef] [Green Version]
- Siu, S.C.; Silversides, C.K. Bicuspid aortic valve disease. J. Am. Coll. Cardiol. 2010, 55, 2789–2800. [Google Scholar] [CrossRef] [Green Version]
- Combs, M.D.; Yutzey, K.E. Heart valve development: Regulatory networks in development and disease. Circ. Res. 2009, 105, 408–421. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Siddiqui, M.A. Signal transduction in early heart development (I): Cardiogenic induction and heart tube formation. Exp. Biol. Med. 2007, 232, 852–865. [Google Scholar]
- Haack, T.; Abdelilah-Seyfried, S. The force within: Endocardial development, mechanotransduction and signalling during cardiac morphogenesis. Development 2016, 143, 373–386. [Google Scholar] [CrossRef] [Green Version]
- Fish, J.E.; Wythe, J.D.; Xiao, T.; Bruneau, B.G.; Stainier, D.Y.; Srivastava, D.; Woo, S. A Slit/miR-218/Robo regulatory loop is required during heart tube formation in zebrafish. Development 2011, 138, 1409–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totong, R.; Schell, T.; Lescroart, F.; Ryckebüsch, L.; Lin, Y.F.; Zygmunt, T.; Herwig, L.; Krudewig, A.; Gershoony, D.; Belting, H.G.; et al. The novel transmembrane protein Tmem2 is essential for coordination of myocardial and endocardial morphogenesis. Development 2011, 138, 4199–4205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinh, L.A.; Stainier, D.Y. Fibronectin regulates epithelial organization during myocardial migration in zebrafish. Dev. Cell 2004, 6, 371–382. [Google Scholar] [CrossRef] [Green Version]
- Bloomekatz, J.; Singh, R.; Prall, O.W.; Dunn, A.C.; Vaughan, M.; Loo, C.S.; Harvey, R.P.; Yelon, D. Platelet-derived growth factor (PDGF) signaling directs cardiomyocyte movement toward the midline during heart tube assembly. Elife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- George, E.L.; Baldwin, H.S.; Hynes, R.O. Fibronectins are essential for heart and blood vessel morphogenesis but are dispensable for initial specification of precursor cells. Blood 1997, 90, 3073–3081. [Google Scholar] [CrossRef]
- Holtzman, N.G.; Schoenebeck, J.J.; Tsai, H.J.; Yelon, D. Endocardium is necessary for cardiomyocyte movement during heart tube assembly. Development 2007, 134, 2379–2386. [Google Scholar] [CrossRef] [Green Version]
- Patra, C.; Diehl, F.; Ferrazzi, F.; van Amerongen, M.J.; Novoyatleva, T.; Schaefer, L.; Mühlfeld, C.; Jungblut, B.; Engel, F.B. Nephronectin regulates atrioventricular canal differentiation via Bmp4-Has2 signaling in zebrafish. Development 2011, 38, 4499–4509. [Google Scholar] [CrossRef] [Green Version]
- Steed, E.; Faggianelli, N.; Roth, S.; Ramspacher, C.; Concordet, J.-P.; Vermot, J. Klf2a Couples Mechanotransduction and Zebrafish Valve Morphogenesis through Fibronectin Synthesis. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Astrof, S.; Crowley, D.; Hynes, R.O. Multiple cardiovascular defects caused by the absence of alternatively spliced segments of fibronectin. Dev. Biol. 2007, 311, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Bakkers, J. Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc. Res. 2011, 91, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Scherz, P.J.; Huisken, J.; Sahai-Hernandez, P.; Stainier, D.Y. High-speed imaging of developing heart valves reveals interplay of morphogenesis and function. Development 2008, 135, 1179–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pestel, J.; Ramadass, R.; Gauvrit, S.; Helker, C.; Herzog, W.; Stainier, D.Y.R. Real-time 3D Visualization of Cellular Rearrangements during Cardiac Valve Formation. Development 2016, 143, 2217–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Wijk, B.; Moorman, A.F.; van den Hoff, M.J. Role of bone morphogenetic proteins in cardiac differentiation. Cardiovasc. Res. 2007, 74, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Sugi, Y.; Yamamura, H.; Okagawa, H.; Markwald, R.R. Bone morphogenetic protein-2 can mediate myocardial regulation of atrioventricular cushion mesenchymal cell formation in mice. Dev. Biol. 2004, 269, 505–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Lu, M.F.; Schwartz, R.J.; Martin, J.F. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development 2005, 132, 5601–5611. [Google Scholar] [CrossRef] [Green Version]
- Palencia-Desai, S.; Rost, M.S.; Schumacher, J.A.; Ton, Q.V.; Craig, M.P.; Baltrunaite, K.; Koenig, A.L.; Wang, J.; Poss, K.D.; Chi, N.C.; et al. Myocardium and BMP signaling are required for endocardial differentiation. Development 2015, 142, 2304–2315. [Google Scholar] [CrossRef] [Green Version]
- Walsh, E.C.; Stainier, D.Y. UDP-glucose dehydrogenase required for cardiac valve formation in zebrafish. Science 2001, 293, 1670–1673. [Google Scholar] [CrossRef]
- Yuasa, S.; Fukuda, K. Multiple roles for BMP signaling in cardiac development. Drug Discov. Today Ther. Strateg. 2008, 5, 209–214. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, B.; Chamberlain, A.A.; Lui, W.; Koirala, P.; Susztak, K.; Klein, D.; Taylor, V.; Zhou, B. Endocardial to myocardial notch-wnt-bmp axis regulates early heart valve development. PLoS ONE 2013, 8, e60244. [Google Scholar] [CrossRef] [Green Version]
- Verhoeven, M.C.; Haase, C.; Christoffels, V.M.; Weidinger, G.; Bakkers, J. Wnt signaling regulates atrioventricular canal formation upstream of BMP and Tbx2. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 435–440. [Google Scholar] [CrossRef]
- Galvin, K.M.; Donovan, M.J.; Lynch, C.A.; Meyer, R.I.; Paul, R.J.; Lorenz, J.N.; Fairchild-Huntress, V.; Dixon, K.L.; Dunmore, J.H.; Gimbrone, M.A.; et al. A role for smad6 in development and homeostasis of the cardiovascular system. Nat. Genet. 2000, 24, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Sugi, Y.; Kern, M.J.; Markwald, R.R.; Burnside, J.L. Periostin Expression is Altered in Aortic Valves in Smad6 Mutant Mice. J. Neonatal. Biol. 2012, 1. [Google Scholar] [CrossRef]
- Hulin, A.; Hortells, L.; Gomez-Stallons, M.V.; O’Donnell, A.; Chetal, K.; Adam, M.; Lancellotti, P.; Oury, C.; Potter, S.S.; Salomonis, N.; et al. Maturation of heart valve cell populations during postnatal remodeling. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowen, C.J.; Zhou, J.; Sung, D.C.; Butcher, J.T. Cadherin-11 coordinates cellular migration and extracellular matrix remodeling during aortic valve maturation. Dev. Biol. 2015, 407, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Gunawan, F.; Gentile, A.; Gauvrit, S.; Stainier, D.; Bensimon-Brito, A. Nfatc1 Promotes Interstitial Cell Formation During Cardiac Valve Development in Zebrafish. Circ. Res. 2020. [Google Scholar] [CrossRef]
- Gallina, D.; Lincoln, J. Dynamic Expression Profiles of Sox9 in Embryonic, Post Natal, and Adult Heart Valve Cell Populations. Anat. Rec. 2019, 302, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Lincoln, J.; Kist, R.; Scherer, G.; Yutzey, K.E. Sox9 is required for precursor cell expansion and extracellular matrix organization during mouse heart valve development. Dev. Biol. 2007, 305, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Plein, A.; Fantin, A.; Ruhrberg, C. Neural crest cells in cardiovascular development. Curr. Top. Dev. Biol. 2015, 111, 183–200. [Google Scholar] [CrossRef]
- Jain, R.; Engleka, K.A.; Rentschler, S.L.; Manderfield, L.J.; Li, L.; Yuan, L.; Epstein, J.A. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J. Clin. Investig. 2011, 121, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Ma, P.; Gu, S.; Karunamuni, G.H.; Jenkins, M.W.; Watanabe, M.; Rollins, A.M. Cardiac neural crest ablation results in early endocardial cushion and hemodynamic flow abnormalities. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1150–H1159. [Google Scholar] [CrossRef] [Green Version]
- Keyte, A.; Hutson, M.R. The neural crest in cardiac congenital anomalies. Differentiation 2012, 84, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Koenig, S.N.; Bosse, K.; Majumdar, U.; Bonachea, E.M.; Radtke, F.; Garg, V. Endothelial Notch1 Is Required for Proper Development of the Semilunar Valves and Cardiac Outflow Tract. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Phillips, H.M.; Mahendran, P.; Singh, E.; Anderson, R.H.; Chaudhry, B.; Henderson, D.J. Neural crest cells are required for correct positioning of the developing outflow cushions and pattern the arterial valve leaflets. Cardiovasc. Res. 2013, 99, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Manderfield, L.J.; Aghajanian, H.; Engleka, K.A.; Lim, L.Y.; Liu, F.; Jain, R.; Li, L.; Olson, E.N.; Epstein, J.A. Hippo signaling is required for Notch-dependent smooth muscle differentiation of neural crest. Development 2015, 142, 2962–2971. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chang, J.Y.; Huang, Y.; Lin, X.; Luo, Y.; Schwartz, R.J.; Martin, J.F.; Wang, F. The FGF-BMP signaling axis regulates outflow tract valve primordium formation by promoting cushion neural crest cell differentiation. Circ. Res. 2010, 107, 1209–1219. [Google Scholar] [CrossRef] [Green Version]
- Odelin, G.; Faure, E.; Coulpier, F.; Di Bonito, M.; Bajolle, F.; Studer, M.; Avierinos, J.F.; Charnay, P.; Topilko, P.; Zaffran, S. Krox20 defines a subpopulation of cardiac neural crest cells contributing to arterial valves and bicuspid aortic valve. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Oomen, P.J.A.; Holland, M.A.; Bouten, C.V.C.; Kuhl, E.; Loerakker, S. Growth and remodeling play opposing roles during postnatal human heart valve development. Sci. Rep. 2018, 8, 1235. [Google Scholar] [CrossRef] [Green Version]
- Pagnozzi, L.A.; Butcher, J.T. Mechanotransduction Mechanisms in Mitral Valve Physiology and Disease Pathogenesis. Front. Cardiovasc. Med. 2017, 4, 83. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Rivkees, S.A. Programmed cell death in the developing heart: Regulation by BMP4 and FGF2. Dev. Dyn. 2000, 217, 388–400. [Google Scholar] [CrossRef]
- Lindsey, S.E.; Butcher, J.T.; Yalcin, H.C. Mechanical regulation of cardiac development. Front. Physiol. 2014, 5, 318. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Zhang, X.; Bu, H.; Liu, N.; Li, H.; Guan, X.; Yan, H.; Wang, Y.; Zhang, H.; Ding, Y.; et al. Shear stress-mediated changes in the expression of complement regulatory protein CD59 on human endothelial progenitor cells by ECM-integrinα. Biochem. Biophys. Res. Commun. 2017, 494, 416–421. [Google Scholar] [CrossRef]
- Sathanoori, R.; Rosi, F.; Gu, B.J.; Wiley, J.S.; Müller, C.E.; Olde, B.; Erlinge, D. Shear stress modulates endothelial KLF2 through activation of P2X4. Purinergic. Signal. 2015, 11, 139–153. [Google Scholar] [CrossRef] [Green Version]
- Hove, J.R.; Köster, R.W.; Forouhar, A.S.; Acevedo-Bolton, G.; Fraser, S.E.; Gharib, M. Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature 2003, 421, 172–177. [Google Scholar] [CrossRef]
- Groenendijk, B.C.; Van der Heiden, K.; Hierck, B.P.; Poelmann, R.E. The role of shear stress on ET-1, KLF2, and NOS-3 expression in the developing cardiovascular system of chicken embryos in a venous ligation model. Physiology 2007, 22, 380–389. [Google Scholar] [CrossRef]
- Goddard, L.M.; Duchemin, A.L.; Ramalingan, H.; Wu, B.; Chen, M.; Bamezai, S.; Yang, J.; Li, L.; Morley, M.P.; Wang, T.; et al. Hemodynamic Forces Sculpt Developing Heart Valves through a KLF2-WNT9B Paracrine Signaling Axis. Dev. Cell 2017, 43, 274–289.e275. [Google Scholar] [CrossRef]
- Vermot, J.; Forouhar, A.; Liebling, M.; Wu, D.; Plummer, D.; Gharib, M. Reversing Blood Flows Act through klf2a to Ensure Normal Valvulogenesis in the Developing Heart. PLoS Biol. 2009, 7, e1000246. [Google Scholar] [CrossRef] [Green Version]
- Dekker, R.J.; Boon, R.A.; Rondaij, M.G.; Kragt, A.; Volger, O.L.; Elderkamp, Y.W.; Meijers, J.C.; Voorberg, J.; Pannekoek, H.; Horrevoets, A.J. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood 2006, 107, 4354–4363. [Google Scholar] [CrossRef] [Green Version]
- Heckel, E.; Boselli, F.; Roth, S.; Krudewig, A.; Belting, H.-G.; Charvin, G.; Vermot, J. Oscillatory Flow Modulates Mechanosensitive klf2a Expression through trpv4 and trpp2 during Heart Valve Development. Curr. Biol. 2015, 25, 1354–1361. [Google Scholar] [CrossRef] [Green Version]
- Donat, S.; Lourenço, M.; Paolini, A.; Otten, C.; Renz, M.; Abdelilah-Seyfried, S. Heg1 and Ccm1/2 proteins control endocardial mechanosensitivity during zebrafish valvulogenesis. Elife 2018, 7. [Google Scholar] [CrossRef]
- Marshall, W.F.; Nonaka, S. Cilia: Tuning in to the cell’s antenna. Curr. Biol. 2006, 16, R604–R614. [Google Scholar] [CrossRef] [Green Version]
- Pala, R.; Jamal, M.; Alshammari, Q.; Nauli, S.M. The Roles of Primary Cilia in Cardiovascular Diseases. Cells 2018, 7, 233. [Google Scholar] [CrossRef] [Green Version]
- Nonaka, S.; Tanaka, Y.; Okada, Y.; Takeda, S.; Harada, A.; Kanai, Y.; Kido, M.; Hirokawa, N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell 1998, 95, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Slough, J.; Cooney, L.; Brueckner, M. Monocilia in the embryonic mouse heart imply a direct role for cilia in cardiac morphogenesis. Dev. Dyn. 2008, 237, 2304–2314. [Google Scholar] [CrossRef]
- Samsa, L.A.; Givens, C.; Tzima, E.; Stainier, D.Y.; Qian, L.; Liu, J. Cardiac contraction activates endocardial Notch signaling to modulate chamber maturation in zebrafish. Development 2015, 142, 4080–4091. [Google Scholar] [CrossRef] [Green Version]
- Fulmer, D.; Toomer, K.; Guo, L.; Moore, K.; Glover, J.; Moore, R.; Stairley, R.; Lobo, G.; Zuo, X.; Dang, Y.; et al. Defects in the Exocyst-Cilia Machinery Cause Bicuspid Aortic Valve Disease and Aortic Stenosis. Circulation 2019, 140, 1331–1341. [Google Scholar] [CrossRef]
- Ten Dijke, P.; Egorova, A.D.; Goumans, M.J.; Poelmann, R.E.; Hierck, B.P. TGF-β signaling in endothelial-to-mesenchymal transition: The role of shear stress and primary cilia. Sci. Signal. 2012, 5. [Google Scholar] [CrossRef]
- Kumar, S.; Kim, C.W.; Simmons, R.D.; Jo, H. Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: Mechanosensitive athero-miRs. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2206–2216. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Williams, D.; Sur, S.; Wang, J.Y.; Jo, H. Role of flow-sensitive microRNAs and long noncoding RNAs in vascular dysfunction and atherosclerosis. Vascul. Pharmacol. 2019, 114, 76–92. [Google Scholar] [CrossRef]
- Rathan, S.; Ankeny, C.J.; Arjunon, S.; Ferdous, Z.; Kumar, S.; Fernandez Esmerats, J.; Heath, J.M.; Nerem, R.M.; Yoganathan, A.P.; Jo, H. Identification of side- and shear-dependent microRNAs regulating porcine aortic valve pathogenesis. Sci. Rep. 2016, 6, 25397. [Google Scholar] [CrossRef] [Green Version]
- Toshima, T.; Watanabe, T.; Narumi, T.; Otaki, Y.; Shishido, T.; Aono, T.; Goto, J.; Watanabe, K.; Sugai, T.; Takahashi, T.; et al. Therapeutic inhibition of microRNA-34a ameliorates aortic valve calcification via modulation of Notch1-Runx2 signaling. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef]
- Sabatino, J.; Wicik, Z.; De Rosa, S.; Eyileten, C.; Jakubik, D.; Spaccarotella, C.; Mongiardo, A.; Postula, M.; Indolfi, C. MicroRNAs fingerprint of bicuspid aortic valve. J. Mol. Cell. Cardiol. 2019, 134, 98–106. [Google Scholar] [CrossRef]
- Banjo, T.; Grajcarek, J.; Yoshino, D.; Osada, H.; Miyasaka, K.Y.; Kida, Y.S.; Ueki, Y.; Nagayama, K.; Kawakami, K.; Matsumoto, T.; et al. Haemodynamically dependent valvulogenesis of zebrafish heart is mediated by flow-dependent expression of miR-21. Nat. Commun. 2013, 4, 1978. [Google Scholar] [CrossRef] [Green Version]
- Pappano, A.J.; Wier, W.G. 10—Control of Cardiac Output: Coupling of Heart and Blood Vessels. In Cardiovascular Physiology, 10th ed.; Elsevier: Philadelphia, PA, USA, 2013; pp. 195–222. [Google Scholar]
- Phoon, C.K. Circulatory physiology in the developing embryo. Curr. Opin. Pediatr. 2001, 13, 456–464. [Google Scholar] [CrossRef]
- Johnson, B.; Garrity, D.; Dasi, P. The transitional cardiac pumping mechanics in the embryonic heart. Cardiovasc. Eng. Technol. 2013, 4, 246–255. [Google Scholar] [CrossRef]
- Forouhar, A.S.; Liebling, M.; Hickerson, A.; Nasiraei-Moghaddam, A.; Tsai, H.J.; Hove, J.R.; Fraser, S.E.; Dickinson, M.E.; Gharib, M. The embryonic vertebrate heart tube is a dynamic suction pump. Science 2006, 312, 751–753. [Google Scholar] [CrossRef] [Green Version]
- Bark, D.L.; Johnson, B.; Garrity, D.; Dasi, L.P. Valveless pumping mechanics of the embryonic heart during cardiac looping: Pressure and flow through micro-PIV. J. Biomech. 2017, 50, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Yalcin, H.C.; Shekhar, A.; McQuinn, T.C.; Butcher, J.T. Hemodynamic patterning of the avian atrioventricular valve. Dev. Dyn. 2011, 240, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Peyronnet, R.; Nerbonne, J.M.; Kohl, P. Cardiac Mechano-Gated Ion Channels and Arrhythmias. Circ. Res. 2016, 118, 311–329. [Google Scholar] [CrossRef] [Green Version]
- Israeli-Rosenberg, S.; Manso, A.M.; Okada, H.; Ross, R.S. Integrins and integrin-associated proteins in the cardiac myocyte. Circ. Res. 2014, 114, 572–586. [Google Scholar] [CrossRef] [Green Version]
- LaCombe, P.; Lappin, S. Physiology, Afterload Reduction; StatPearls Publishing LLC.: Tresasure Island, FL, USA, 2019. [Google Scholar]
- Krüger, M.; Kötter, S. Titin, a Central Mediator for Hypertrophic Signaling, Exercise-Induced Mechanosignaling and Skeletal Muscle Remodeling. Front. Physiol. 2016, 7, 76. [Google Scholar] [CrossRef] [Green Version]
- Burridge, K. Focal adhesions: A personal perspective on a half century of progress. FEBS J. 2017, 284, 3355–3361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davila, C.D.; Forfia, P.R. Management of Severe Pulmonary Hypertension in Patients Undergoing Mitral Valve Surgery. Curr. Treat. Options Cardiovasc. Med. 2015, 17, 26. [Google Scholar] [CrossRef]
- Hirt, M.N.; Sörensen, N.A.; Bartholdt, L.M.; Boeddinghaus, J.; Schaaf, S.; Eder, A.; Vollert, I.; Stöhr, A.; Schulze, T.; Witten, A.; et al. Increased afterload induces pathological cardiac hypertrophy: A new in vitro model. Basic Res. Cardiol. 2012, 107, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, D.; Larson, M.G.; Vasan, R.S.; Kannel, W.B.; Ho, K.K. The progression from hypertension to congestive heart failure. JAMA 1996, 275, 1557–1562. [Google Scholar] [CrossRef]
- Rahimi, K.; Mohseni, H.; Kiran, A.; Tran, J.; Nazarzadeh, M.; Rahimian, F.; Woodward, M.; Dwyer, T.; MacMahon, S.; Otto, C.M. Elevated blood pressure and risk of aortic valve disease: A cohort analysis of 5.4 million UK adults. Eur. Heart J. 2018, 39, 3596–3603. [Google Scholar] [CrossRef]
- Rahimi, K.; Mohseni, H.; Otto, C.M.; Conrad, N.; Tran, J.; Nazarzadeh, M.; Woodward, M.; Dwyer, T.; MacMahon, S. Elevated blood pressure and risk of mitral regurgitation: A longitudinal cohort study of 5.5 million United Kingdom adults. PLoS Med. 2017, 14, e1002404. [Google Scholar] [CrossRef]
- Pant, A.D.; Thomas, V.S.; Black, A.L.; Verba, T.; Lesicko, J.G.; Amini, R. Pressure-induced microstructural changes in porcine tricuspid valve leaflets. Acta Biomater. 2018, 67, 248–258. [Google Scholar] [CrossRef]
- Balachandran, K.; Konduri, S.; Sucosky, P.; Jo, H.; Yoganathan, A.P. An ex vivo study of the biological properties of porcine aortic valves in response to circumferential cyclic stretch. Ann. Biomed. Eng. 2006, 34, 1655–1665. [Google Scholar] [CrossRef] [Green Version]
- Warnock, J.N.; Nanduri, B.; Pregonero Gamez, C.A.; Tang, J.; Koback, D.; Muir, W.M.; Burgess, S.C. Gene Profiling of Aortic Valve Interstitial Cells under Elevated Pressure Conditions: Modulation of Inflammatory Gene Networks. Int. J. Inflam. 2011, 2011, 176412. [Google Scholar] [CrossRef] [Green Version]
- Amstrup Funder, J.; Christian Danielsen, C.; Baandrup, U.; Martin Bibby, B.; Carl Andelius, T.; Toft Brøndum, E.; Wang, T.; Michael Hasenkam, J. How Heart Valves Evolve to Adapt to an Extreme-Pressure System: Morphologic and Biomechanical Properties of Giraffe Heart Valves. J. Heart Valve Dis. 2017, 26, 63–71. [Google Scholar]
- Balachandran, K.; Sucosky, P.; Jo, H.; Yoganathan, A.P. Elevated cyclic stretch induces aortic valve calcification in a bone morphogenic protein-dependent manner. Am. J. Pathol. 2010, 177, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.A.; Masters, K.S.; Shah, D.N.; Anseth, K.S.; Leinwand, L.A. Valvular myofibroblast activation by transforming growth factor-beta: Implications for pathological extracellular matrix remodeling in heart valve disease. Circ. Res. 2004, 95, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández Esmerats, J.; Heath, J.; Jo, H. Shear-Sensitive Genes in Aortic Valve Endothelium. Antioxid. Redox Signal. 2016, 25, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midgett, M.; Rugonyi, S. Congenital heart malformations induced by hemodynamic altering surgical interventions. Front. Physiol. 2014, 5, 287. [Google Scholar] [CrossRef] [Green Version]
- Sedmera, D.; Pexieder, T.; Rychterova, V.; Hu, N.; Clark, E.B. Remodeling of chick embryonic ventricular myoarchitecture under experimentally changed loading conditions. Anat. Rec. 1999, 254, 238–252. [Google Scholar] [CrossRef]
- Freud, L.R.; McElhinney, D.B.; Marshall, A.C.; Marx, G.R.; Friedman, K.G.; del Nido, P.J.; Emani, S.M.; Lafranchi, T.; Silva, V.; Wilkins-Haug, L.E.; et al. Fetal aortic valvuloplasty for evolving hypoplastic left heart syndrome: Postnatal outcomes of the first 100 patients. Circulation 2014, 130, 638–645. [Google Scholar] [CrossRef] [Green Version]
- Midgett, M.; López, C.S.; David, L.; Maloyan, A.; Rugonyi, S. Increased Hemodynamic Load in Early Embryonic Stages Alters Endocardial to Mesenchymal Transition. Front. Physiol. 2017, 8, 56. [Google Scholar] [CrossRef] [Green Version]
- Pang, K.L.; Parnall, M.; Loughna, S. Effect of altered haemodynamics on the developing mitral valve in chick embryonic heart. J. Mol. Cell. Cardiol. 2017, 108, 114–126. [Google Scholar] [CrossRef]
- Menon, V.; Eberth, J.F.; Goodwin, R.L.; Potts, J.D. Altered Hemodynamics in the Embryonic Heart Affects Outflow Valve Development. J. Cardiovasc. Dev. Dis. 2015, 2, 108–124. [Google Scholar] [CrossRef]
- Pesevski, Z.; Kvasilova, A.; Stopkova, T.; Nanka, O.; Drobna Krejci, E.; Buffinton, C.; Kockova, R.; Eckhardt, A.; Sedmera, D. Endocardial Fibroelastosis is Secondary to Hemodynamic Alterations in the Chick Embryonic Model of Hypoplastic Left Heart Syndrome. Dev. Dyn. 2018, 247, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Bustelo, X.R.; Sauzeau, V.; Berenjeno, I.M. GTP-binding proteins of the Rho/Rac family: Regulation, effectors and functions in vivo. Bioessays 2007, 29, 356–370. [Google Scholar] [CrossRef] [Green Version]
- Parri, M.; Chiarugi, P. Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. 2010, 8, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, R.A.; Yalcin, H.C.; MacKay, J.L.; Sauls, K.; Norris, R.; Kumar, S.; Butcher, J.T. Cyclic Mechanical Loading Is Essential for Rac1-Mediated Elongation and Remodeling of the Embryonic Mitral Valve. Curr. Biol. 2016, 26, 27–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, H.; Biechler, S.; Junor, L.; Yost, M.J.; Dean, D.; Li, J.; Potts, J.D.; Goodwin, R.L. Fluid flow forces and rhoA regulate fibrous development of the atrioventricular valves. Dev. Biol. 2013, 374, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Masters, K.S. Role of the Rho pathway in regulating valvular interstitial cell phenotype and nodule formation. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H448–H458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilluy, C.; Swaminathan, V.; Garcia-Mata, R.; O’Brien, E.T.; Superfine, R.; Burridge, K. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat. Cell Biol. 2011, 13, 722–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lessey, E.C.; Guilluy, C.; Burridge, K. From mechanical force to RhoA activation. Biochemistry 2012, 51, 7420–7432. [Google Scholar] [CrossRef]
- Parra, V.; Rothermel, B.A. Calcineurin signaling in the heart: The importance of time and place. J. Mol. Cell. Cardiol. 2017, 103, 121–136. [Google Scholar] [CrossRef] [Green Version]
- Heineke, J.; Ruetten, H.; Willenbockel, C.; Gross, S.C.; Naguib, M.; Schaefer, A.; Kempf, T.; Hilfiker-Kleiner, D.; Caroni, P.; Kraft, T.; et al. Attenuation of cardiac remodeling after myocardial infarction by muscle LIM protein-calcineurin signaling at the sarcomeric Z-disc. Proc. Natl. Acad. Sci. USA 2005, 102, 1655–1660. [Google Scholar] [CrossRef] [Green Version]
- Jeong, D.; Kim, J.M.; Cha, H.; Oh, J.G.; Park, J.; Yun, S.H.; Ju, E.S.; Jeon, E.S.; Hajjar, R.J.; Park, W.J. PICOT attenuates cardiac hypertrophy by disrupting calcineurin-NFAT signaling. Circ. Res. 2008, 102, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Frank, D.; Frey, N. Cardiac Z-disc signaling network. J. Biol. Chem. 2011, 286, 9897–9904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.P.; Neilson, J.R.; Bayle, J.H.; Gestwicki, J.E.; Kuo, A.; Stankunas, K.; Graef, I.A.; Crabtree, G.R. A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell 2004, 118, 649–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Zheng, Y.; Liu, X.; Yan, L.; Fan, X.; Yong, J.; Hu, Y.; Dong, J.; Li, Q.; Wu, X.; et al. Single-Cell Transcriptome Analysis Maps the Developmental Track of the Human Heart. Cell Rep. 2019, 26, 1934–1950.e1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnette, D.N.; VandeKopple, M.; Wu, Y.; Willoughby, D.A.; Lincoln, J. RNA-seq analysis to identify novel roles of scleraxis during embryonic mouse heart valve remodeling. PLoS ONE 2014, 9, e101425. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahuja, N.; Ostwald, P.; Bark, D.; Garrity, D. Biomechanical Cues Direct Valvulogenesis. J. Cardiovasc. Dev. Dis. 2020, 7, 18. https://doi.org/10.3390/jcdd7020018
Ahuja N, Ostwald P, Bark D, Garrity D. Biomechanical Cues Direct Valvulogenesis. Journal of Cardiovascular Development and Disease. 2020; 7(2):18. https://doi.org/10.3390/jcdd7020018
Chicago/Turabian StyleAhuja, Neha, Paige Ostwald, David Bark, and Deborah Garrity. 2020. "Biomechanical Cues Direct Valvulogenesis" Journal of Cardiovascular Development and Disease 7, no. 2: 18. https://doi.org/10.3390/jcdd7020018
APA StyleAhuja, N., Ostwald, P., Bark, D., & Garrity, D. (2020). Biomechanical Cues Direct Valvulogenesis. Journal of Cardiovascular Development and Disease, 7(2), 18. https://doi.org/10.3390/jcdd7020018