Transforming Growth Factor Beta3 is Required for Cardiovascular Development


 ,
,  , ,
, , 

Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Mouse Strains
2.3. Histology and Immunohistochemistry
2.4. TUNEL Staining
2.5. Cell Proliferation
2.6. AMIRA Three-Dimensional (3-D) Reconstruction and Volumetric Analysis
2.7. RNAscope In Situ Hybridization
2.8. Collagen Gel Contraction Assays
2.9. Western Blot Analysis
3. Results
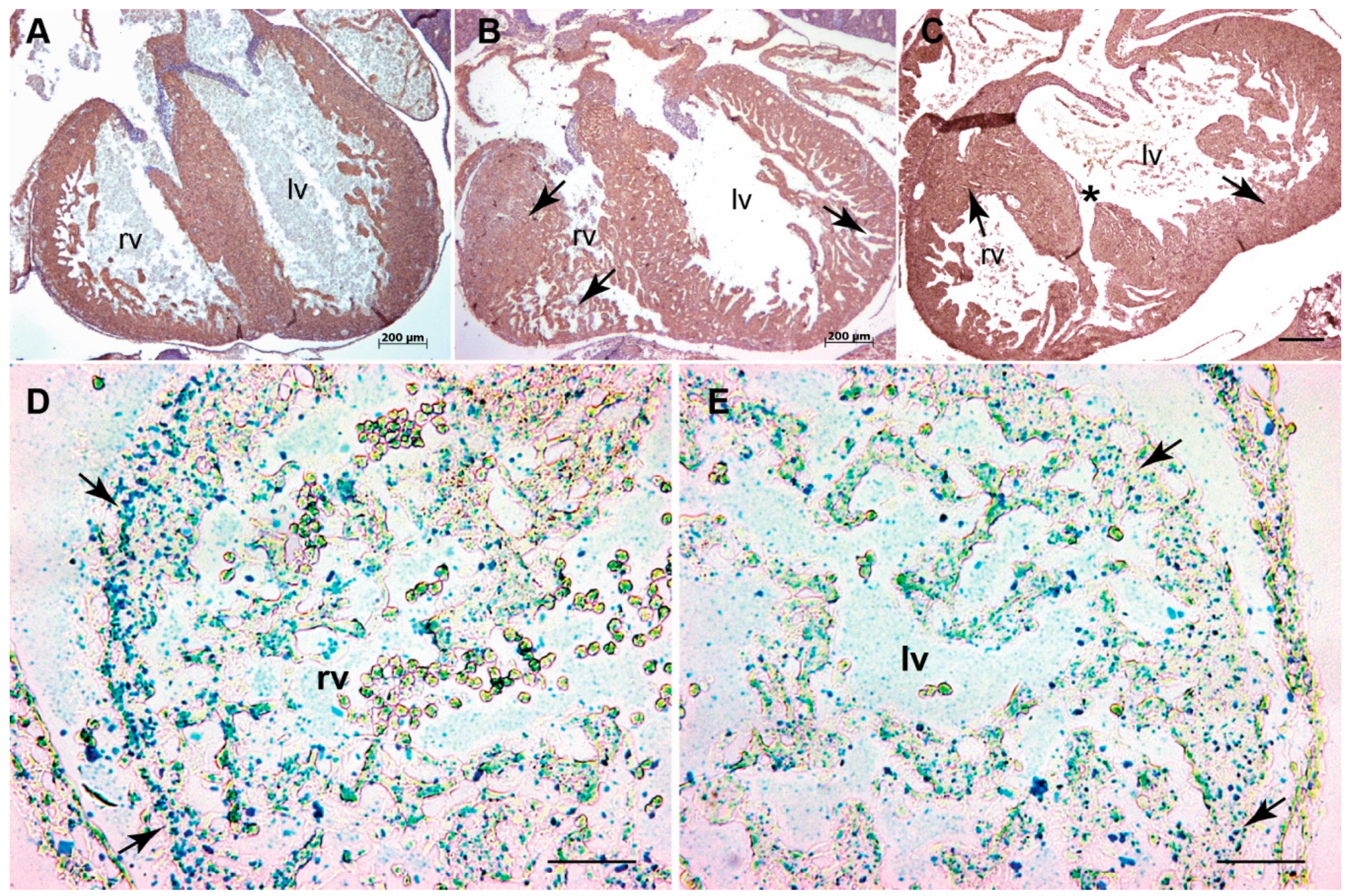
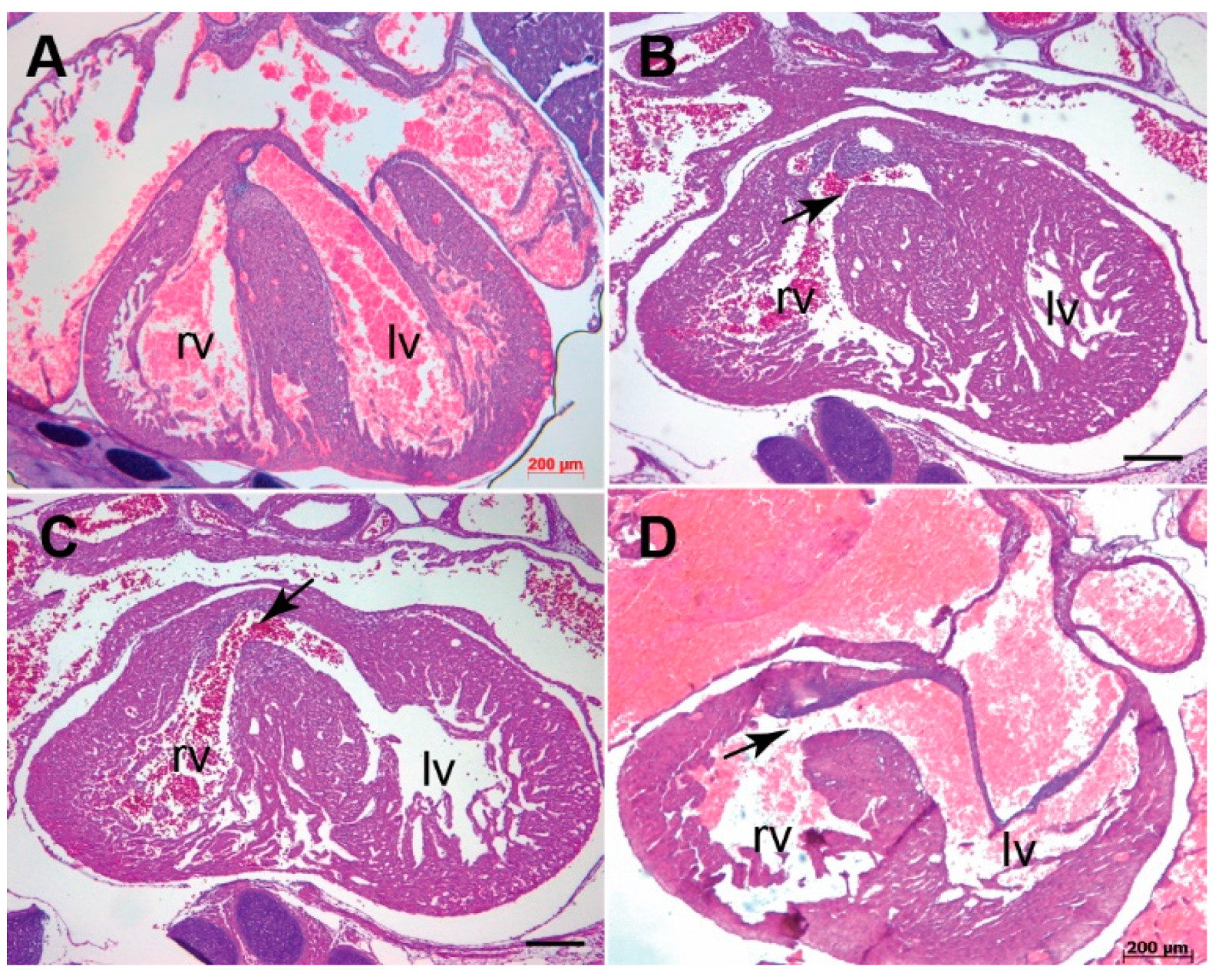
3.1. Systemic Tgfb3 Deletion Disrupts Cardiac Development
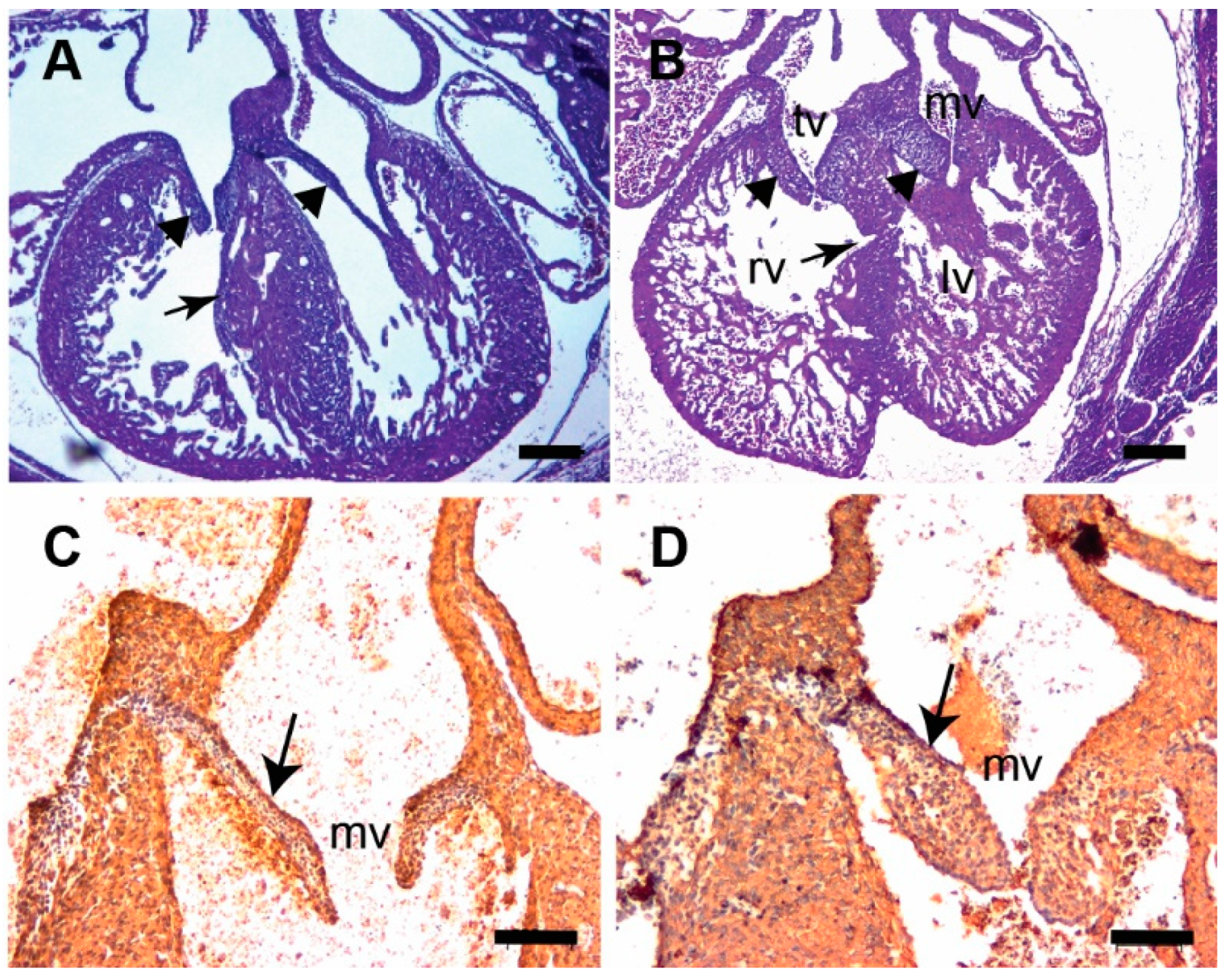
3.2. Tgfb3 Deletion Leads to Outflow Tract Cushion Remodeling and Septal Defects
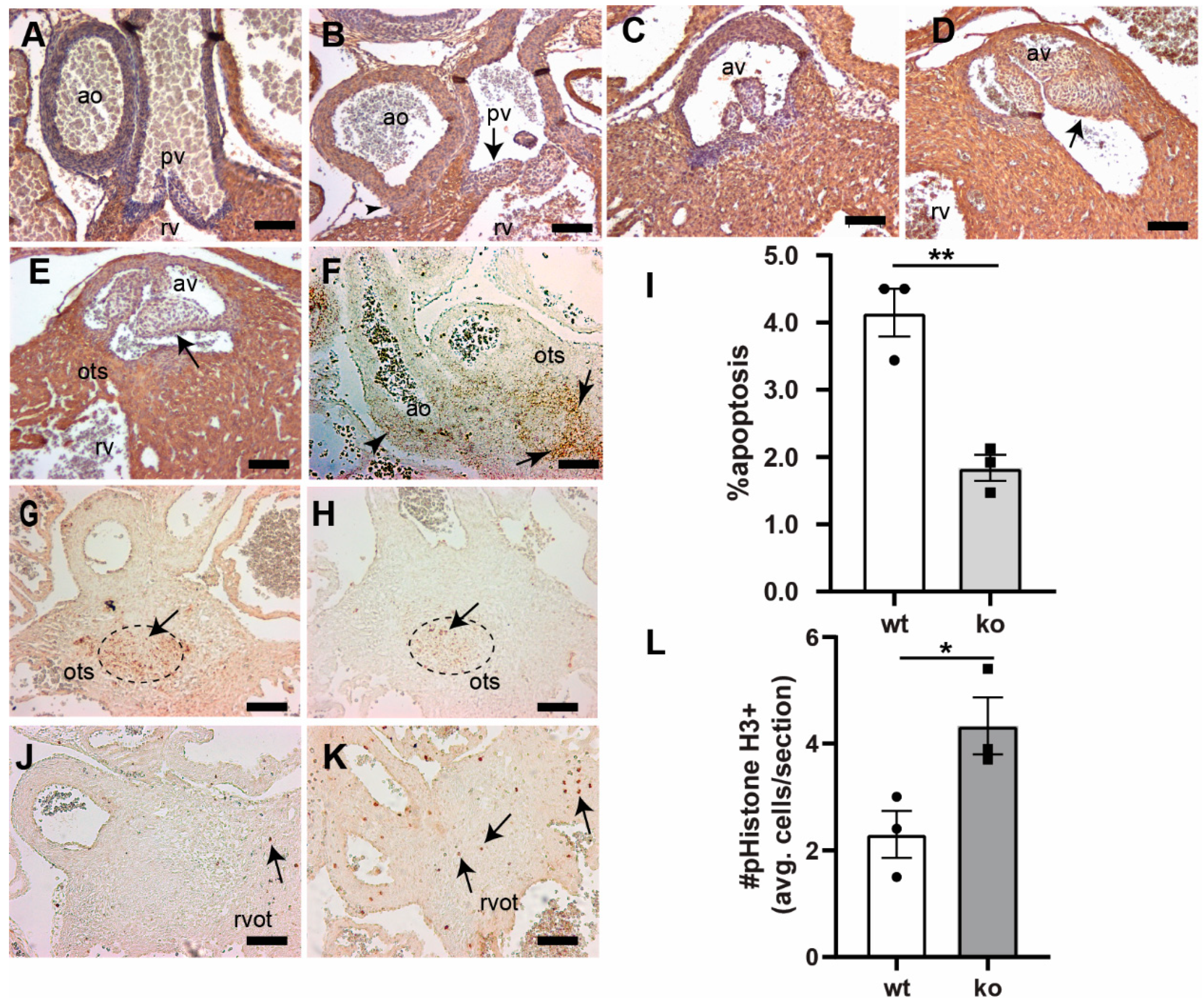
3.3. Tgfb3 Deletion Leads to AV Cushion Remodeling Defects
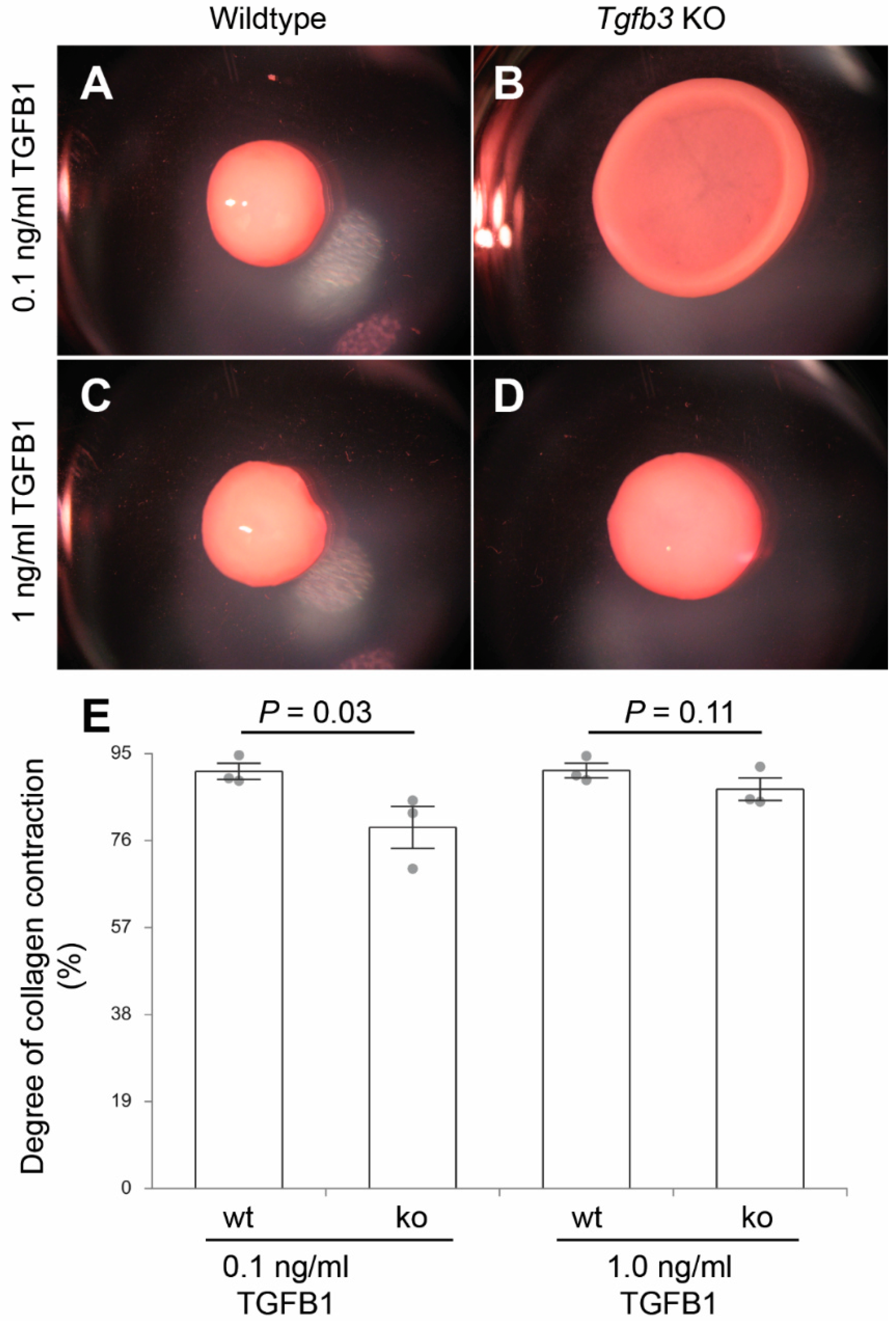
3.4. TGFβ3 Is Required for Extracellular Matrix Reorganization
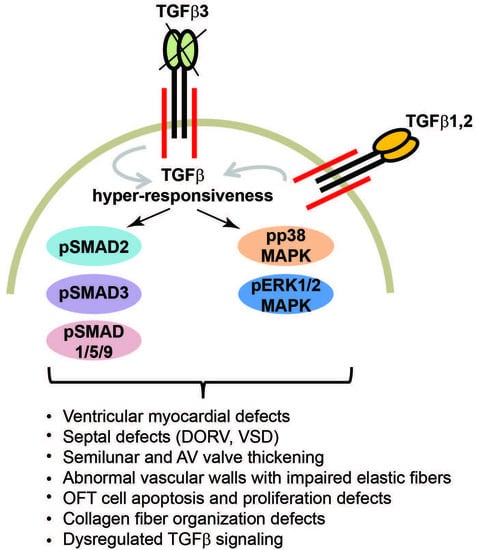
3.5. Tgfb3 Deletion Leads to Hyper-Activated SMAD2/3 and SMAD1/5 Signaling Pathways
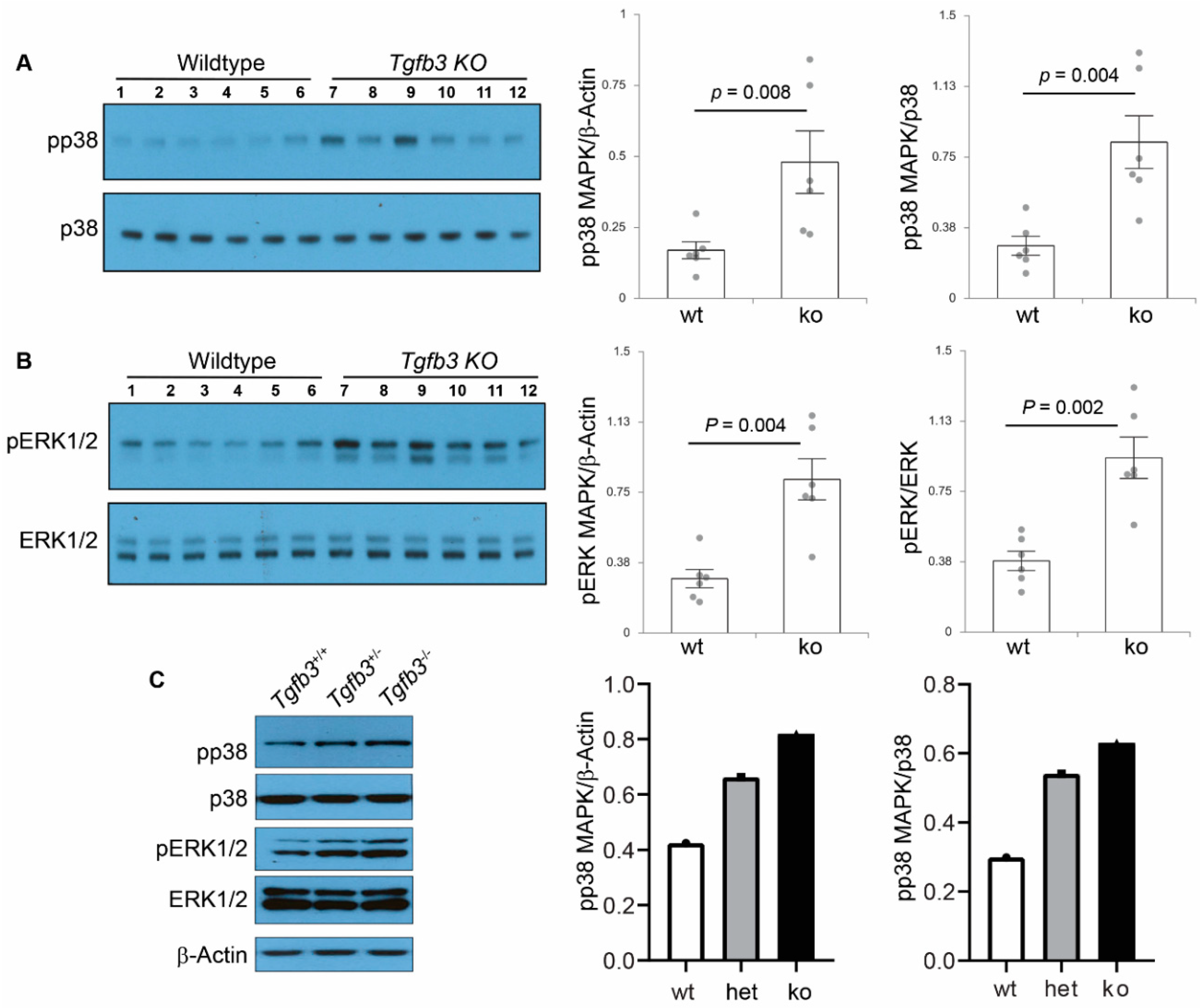
3.6. TGFβ3-Deficiency Triggers Activation of Non-Canonical MAPK-Mediated TGFβ Signaling Pathways
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Proetzel, G.; Pawlowski, S.A.; Wiles, M.V.; Yin, M.; Boivin, G.P.; Howles, P.N.; Ding, J.; Ferguson, M.W.J.; Doetschman, T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 1995, 11, 409–414. [Google Scholar] [CrossRef]
- Kaartinen, V.; Voncken, J.W.; Shuler, C.; Warburton, D.; Bu, D.; Heisterkamp, N.; Groffen, J. Abnormal lung development and cleft palate in mice lacking TGF–β3 indicates defects of epithelial–mesenchymal interaction. Nat. Genet. 1995, 11, 415–421. [Google Scholar] [CrossRef]
- Hu, L.; Liu, J.; Li, Z.; Ozturk, F.; Gurumurthy, C.; Romano, R.-A.; Sinha, S.; Nawshad, A. TGFβ3 Regulates Periderm Removal Through ΔNp63 in the Developing Palate. J. Cell. Physiol. 2015, 230, 1212–1225. [Google Scholar] [CrossRef]
- Chopra, S.; Al-Sammarraie, N.; Lai, Y.; Azhar, M. Increased canonical WNT/β-catenin signalling and myxomatous valve disease. Cardiovasc. Res. 2017, 113, 6–9. [Google Scholar] [CrossRef][Green Version]
- Azhar, M.; Ware, S.M. Genetic and Developmental Basis of Cardiovascular Malformations. Clin. Perinatol. 2016, 43, 39–53. [Google Scholar] [CrossRef]
- Tamargo, J. TGFβ3 mutations cause arrhythmogenic right ventricular dysplasia type 1 and open the door to understanding the biological role of TGFβ3 (where there’s a will, there’s a way). Cardiovasc. Res. 2012, 96, 188–190. [Google Scholar] [CrossRef]
- Beffagna, G.; Occhi, G.; Nava, A.; Vitiello, L.; Ditadi, A.; Basso, C.; Bauce, B.; Carraro, G.; Thiene, G.; Towbin, J.A.; et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 2005, 65, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, Y.; Maruyama, M.; Zhu, W.; Chen, H.; Zhang, W.; Reuter, S.; Lin, S.-F.; Haneline, L.S.; Field, L.J.; et al. Restrictive loss of plakoglobin in cardiomyocytes leads to arrhythmogenic cardiomyopathy. Hum. Mol. Genet. 2011, 20, 4582–4596. [Google Scholar] [CrossRef] [PubMed]
- Bertoli-Avella, A.M.; Gillis, E.; Morisaki, H.; Verhagen, J.M.; De Graaf, B.M.; Van De Beek, G.; Gallo, E.; Kruithof, B.P.; Venselaar, H.; Myers, L.A.; et al. Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 2015, 65, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- Schepers, D.; Tortora, G.; Morisaki, H.; MacCarrick, G.; Lindsay, M.; Liang, D.; Mehta, S.G.; Hague, J.; Verhagen, J.M.; Van De Laar, I.M.; et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum. Mutat. 2018, 39, 621–634. [Google Scholar] [CrossRef]
- Mégarbané, A.; Deepthi, A.; Obeid, M.; Al-Ali, M.T.; Gambarini, A.; El-Hayek, S. Homozygous deletion of exons 2–7 within TGFB3 gene in a child with severe Loeys-Dietz syndrome and Marfan-like features. Am. J. Med. Genet. Part A 2020, 182, 1230–1235. [Google Scholar] [CrossRef]
- Matyas, G.; Naef, P.; Tollens, M.; Oexle, K. De novo mutation of the latency-associated peptide domain of TGFB3 in a patient with overgrowth and Loeys-Dietz syndrome features. Am. J. Med. Genet. Part A 2014, 164, 2141–2143. [Google Scholar] [CrossRef] [PubMed]
- Rienhoff, H.Y.; Yeo, C.-Y.; Morissette, R.; Khrebtukova, I.; Melnick, J.; Luo, S.; Leng, N.; Kim, Y.-J.; Schroth, G.; Westwick, J.; et al. A mutation in TGFB3 associated with a syndrome of low muscle mass, growth retardation, distal arthrogryposis and clinical features overlapping with marfan and loeys–dietz syndrome. Am. J. Med. Genet. Part A 2013, 161, 2040–2046. [Google Scholar] [CrossRef] [PubMed]
- Rienhoff, H.Y. Response to “De novo mutation of the TGFB3 latency-associated peptide domain in a patient with overgrowth and Loeys-Dietz syndrome features”. Am. J. Med. Genet. Part A 2014, 164, 2144–2145. [Google Scholar] [CrossRef]
- Akhurst, R.J. The paradoxical TGF-beta vasculopathies. Nat. Genet. 2012, 44, 838–839. [Google Scholar] [CrossRef] [PubMed]
- Doetschman, T.; Barnett, J.V.; Runyan, R.; Camenisch, T.D.; Heimark, R.L.; Granzier, H.L.; Conway, S.J.; Azhar, M. Transforming growth factor beta signaling in adult cardiovascular diseases and repair. Cell Tissue Res. 2011, 347, 203–223. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J. Mol. Cell Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Boil. 2016, 8, a021873. [Google Scholar] [CrossRef]
- Markwald, R.R.; Norris, R.A.; Moreno-Rodriguez, R.; Levine, R.A. Developmental basis of adult cardiovascular diseases: Valvular heart diseases. Ann. N. Y. Acad. Sci. 2010, 1188, 177–183. [Google Scholar] [CrossRef]
- Goumans, M.J.; Dijke, P.T. TGF-β Signaling in Control of Cardiovascular Function. Cold Spring Harb. Perspect. Boil. 2018, 10, a022210. [Google Scholar] [CrossRef]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Boil. 2016. [Google Scholar] [CrossRef]
- Ramachandran, A.; Vizan, P.; Das, D.; Chakravarty, P.; Vogt, J.; Rogers, K.; Müller, P.; Hinck, A.P.; Sapkota, G.P.; Hill, C.S. TGF-β uses a novel mode of receptor activation to phosphorylate SMAD1/5 and induce epithelial-to-mesenchymal transition. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gudey, S.; Landström, M. Non-Smad signaling pathways. Cell Tissue Res. 2011, 347, 11–20. [Google Scholar] [CrossRef]
- Doetschman, T. Interpretation of phenotype in genetically engineered mice. Lab. Anim. Sci. 1999, 49, 137–143. [Google Scholar] [PubMed]
- Heldin, C.-H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Boil. 2016, 8. [Google Scholar] [CrossRef]
- Molin, D.G.; Bartram, U.; Van der Heiden, K.; Van Iperen, L.; Speer, C.P.; Hierck, B.P.; Poelmann, R.E.; Gittenberger-de-Groot, A.C. Expression patterns of Tgfbeta1-3 associate with myocardialisation of the outflow tract and the development of the epicardium and the fibrous heart skeleton. Dev. Dyn. 2003, 227, 431–444. [Google Scholar] [CrossRef]
- Rampazzo, A.; Beffagna, G.; Nava, A.; Occhi, G.; Bauce, B.; Noiato, M.; Basso, C.; Frigo, G.; Thiene, G.; Towbin, J.; et al. Arrhythmogenic right ventricular cardiomyopathy type 1 (ARVD1): Confirmation of locus assignment and mutation screening of four candidate genes. Eur. J. Hum. Genet. 2003, 11, 69–76. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Frangogiannis, N.G. Fibroblasts and the extracellular matrix in right ventricular disease. Cardiovasc. Res. 2017, 113, 1453–1464. [Google Scholar] [CrossRef]
- Azhar, M.; Brown, K.; Gard, C.; Chen, H.; Rajan, S.; Elliott, D.A.; Stevens, M.V.; Camenisch, T.D.; Conway, S.J.; Doetschman, T. Transforming growth factor Beta2 is required for valve remodeling during heart development. Dev. Dyn. 2011, 240, 2127–2141. [Google Scholar] [CrossRef]
- Menon, V.; Eberth, J.F.; Goodwin, R.L.; Potts, J.D. Altered Hemodynamics in the Embryonic Heart Affects Outflow Valve Development. J. Cardiovasc. Dev. Dis. 2015, 2, 108–124. [Google Scholar] [CrossRef]
- Wang, F.; Flanagan, J.; Su, N.; Wang, L.-C.; Bui, S.; Nielson, A.; Wu, X.; Vo, H.-T.; Ma, X.-J.; Luo, Y. RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 2012, 14, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Todaro, G.J.; Green, H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Boil. 1963, 17, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Lane, B.A.; Harmon, K.A.; Goodwin, R.L.; Yost, M.J.; Shazly, T.; Eberth, J.F. Constitutive modeling of compressible type-I collagen hydrogels. Med. Eng. Phys. 2018, 53, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Haskett, D.; Doyle, J.J.; Gard, C.; Chen, H.; Ball, C.; Estabrook, M.A.; Encinas, A.C.; Dietz, H.C.; Utzinger, U.; Geest, J.P.V.; et al. Altered tissue behavior of a non-aneurysmal descending thoracic aorta in the mouse model of Marfan syndrome. Cell Tissue Res. 2011, 347, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Snider, P.; Hinton, R.B.; Moreno-Rodriguez, R.A.; Wang, J.; Rogers, R.; Lindsley, A.; Li, F.; Ingram, D.A.; Menick, D.; Field, L.; et al. Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circ. Res. 2008, 102, 752–760. [Google Scholar] [CrossRef]
- Poelmann, R.E.; Groot, A.C.G.-D. Apoptosis as an instrument in cardiovascular development. Birth Defects Res. C 2005, 75, 305–313. [Google Scholar] [CrossRef]
- Ferguson, M.W.J. Craniofacial malformations: Towards a molecular understanding. Nat. Genet. 1994, 6, 329–330. [Google Scholar] [CrossRef]
- Loeys, B.L.; Dietz, H.C. Loeys-Dietz Syndrome. In Gene Reviews [Internet]; Adam, M.P., Ardinger, H.H., et al., Eds.; University of Washington: Seattle, WA, USA, 2018; pp. 1993–2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1133/ (accessed on 23 April 2020).
- Bartram, U.; Molin, D.G.; Wisse, L.J.; Mohamad, A.; Sanford, L.P.; Doetschman, T.; Speer, C.P.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in Tgfb2 knockout mice. Circulation 2001, 103, 2745–2752. [Google Scholar] [CrossRef]
- Mercado-Pimentel, M.E.; Runyan, R. Multiple Transforming Growth Factor-β Isoforms and Receptors Function during Epithelial-Mesenchymal Cell Transformation in the Embryonic Heart. Cells Tissues Organs 2007, 185, 146–156. [Google Scholar] [CrossRef]
- Azhar, M.; Runyan, R.; Gard, C.; Sanford, L.P.; Miller, M.L.; Andringa, A.; Pawlowski, S.; Rajan, S.; Doetschman, T.; Doetschman, T. Ligand-specific function of transforming growth factor beta in epithelial-mesenchymal transition in heart development. Dev. Dyn. 2009, 238, 431–442. [Google Scholar] [CrossRef]
- Hoover, L.L.; Burton, E.G.; O’Neill, M.L.; Brooks, B.A.; Sreedharan, S.; Dawson, N.A.; Kubalak, S.W. Retinoids regulate TGFβ signaling at the level of Smad2 phosphorylation and nuclear accumulation. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2008, 1783, 2279–2286. [Google Scholar] [CrossRef] [PubMed]
- Kubalak, S.W.; Hutson, D.R.; Scott, K.K.; Shannon, R.A. Elevated transforming growth factor beta2 enhances apoptosis and contributes to abnormal outflow tract and aortic sac development in retinoic X receptor alpha knockout embryos. Development 2002, 129, 733–746. [Google Scholar] [PubMed]
- Walker, D.L.; Vacha, S.J.; Kirby, M.L.; Lo, C. Connexin43 deficiency causes dysregulation of coronary vasculogenesis. Dev. Boil. 2005, 284, 479–498. [Google Scholar] [CrossRef] [PubMed]
- Guner-Ataman, B.; Paffett-Lugassy, N.; Adams, M.S.; Nevis, K.R.; Jahangiri, L.; Obregon, P.; Kikuchi, K.; Poss, K.D.; Burns, C.E.; Burns, C.G. Zebrafish second heart field development relies on progenitor specification in anterior lateral plate mesoderm and nkx2.5 function. Development 2013, 140, 1353–1363. [Google Scholar] [CrossRef]
- Watanabe, M.; Jafri, A.; Fisher, S.A. Apoptosis Is Required for the Proper Formation of the Ventriculo-Arterial Connections. Dev. Boil. 2001, 240, 274–288. [Google Scholar] [CrossRef]
- Vrljicak, P.; Chang, A.C.Y.; Morozova, O.; Wederell, E.D.; Niessen, K.; Marra, M.A.; Karsan, A.; Hoodless, P. Genomic analysis distinguishes phases of early development of the mouse atrio-ventricular canal. Physiol. Genom. 2009, 40, 150–157. [Google Scholar] [CrossRef]
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; Doyle, J.J.; Gallo, E.; Fert-Bober, J.; Kempers, M.J.; Fishman, E.K.; Chen, Y.; Myers, L.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922–927. [Google Scholar] [CrossRef]
- Dunker, N.; Krieglstein, K. Tgfbeta2−/− Tgfbeta3−/− double knockout mice display severe midline fusion defects and early embryonic lethality. Anat. Embryol. 2002, 206, 73–83. [Google Scholar]
- Hu, B.C.; Li, L.; Sun, R.; Gao, P.; Zhu, D.; Wang, J.-G.; Chu, S.-L. The association between transforming growth factor β3 polymorphisms and left ventricular structure in hypertensive subjects. Clin. Chim. Acta 2010, 411, 558–562. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.G.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.-J.; et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Fix, C.; Carver-Molina, A.; Chakrabarti, M.; Azhar, M.; Carver, W. Effects of the isothiocyanate sulforaphane on TGF-β1-induced rat cardiac fibroblast activation and extracellular matrix interactions. J. Cell. Physiol. 2019, 234, 13931–13941. [Google Scholar] [CrossRef]
- Ng, C.M.; Cheng, A.; Myers, L.A.; Martínez-Murillo, F.; Jie, C.; Bedja, D.; Gabrielson, K.L.; Hausladen, J.M.; Mecham, R.P.; Judge, D.P.; et al. TGF-β–dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J. Clin. Investig. 2004, 114, 1586–1592. [Google Scholar] [CrossRef]
- Murata, H.; Zhou, L.; Ochoa, S.; Hasan, A.; Badiavas, E.; Falanga, V. TGF-β3 Stimulates and Regulates Collagen Synthesis Through TGF-β1-Dependent and Independent Mechanisms. J. Investig. Dermatol. 1997, 108, 258–262. [Google Scholar] [CrossRef]
- Groot, A.C.G.-D.; Calkoen, E.E.; Poelmann, R.E.; Bartelings, M.M.; Jongbloed, M.R.M. Morphogenesis and molecular considerations on congenital cardiac septal defects. Ann. Med. 2014, 46, 640–652. [Google Scholar] [CrossRef]
- Conway, S.J.; Kaartinen, V. TGFbeta superfamily signaling in the neural crest lineage. Cell Adh. Migr. 2011, 5, 232–236. [Google Scholar] [CrossRef]
- Neeb, Z.; Lajiness, J.D.; Bolanis, E.; Conway, S.J. Cardiac outflow tract anomalies. Wiley Interdiscip. Rev. Dev. Boil. 2013, 2, 499–530. [Google Scholar] [CrossRef]
- Groot, A.C.G.-D.; Winter, E.M.; Bartelings, M.M.; Goumans, M.J.; DeRuiter, M.C.; Poelmann, R.E. The arterial and cardiac epicardium in development, disease and repair. Differentiation 2012, 84, 41–53. [Google Scholar] [CrossRef]
- Lockhart, M.M.; Hoff, M.V.D.; Wessels, A. The Role of the Epicardium in the Formation of the Cardiac Valves in the Mouse. In Etiology and Morphogenesis of Congenital Heart Disease; Springer Science and Business Media LLC: Berlin, Germany, 2016; pp. 161–167. [Google Scholar]
- Wessels, A.; Pérez-Pomares, J.M. The epicardium and epicardially derived cells (EPDCs) as cardiac stem cells. Anat. Rec. Part A Discov. Mol. Cell. Evol. Boil. 2003, 276, 43–57. [Google Scholar] [CrossRef]
- Vicente-Steijn, R.; Scherptong, R.W.C.; Kruithof, B.P.T.; Duim, S.N.; Goumans, M.J.T.H.; Wisse, L.J.; Zhou, B.; Pu, W.T.; Poelmann, R.E.; Schalij, M.J.; et al. Regional differences in WT-1 and Tcf21 expression during ventricular development: Implications for myocardial compaction. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Poelmann, R.E.; Groot, A.C.G.-D.; Vicente-Steijn, R.; Wisse, L.J.; Bartelings, M.M.; Everts, S.; Hoppenbrouwers, T.; Kruithof, B.P.T.; Jensen, B.; De Bruin, P.W.; et al. Evolution and Development of Ventricular Septation in the Amniote Heart. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Doetschman, T.; Georgieva, T.; Li, H.; Reed, T.D.; Grisham, C.; Friel, J.; Estabrook, M.A.; Gard, C.; Sanford, L.; Azhar, M. Generation of mice with a conditional allele for the transforming growth factor beta3 gene. Genes 2012, 50, 59–66. [Google Scholar] [CrossRef] [PubMed]
Sample availability: not available. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID# | Genotype | Age | DORV | AV Thickening | PV Thickening | Vascular walls Abnormalities | VSD | AVSD | Thin Myocardium | RV Hyperplasia | MV Thickening | TV Thickening |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KO1 | Tgfb3−/− | e16.5-17.5 | No | No | No | No | No | No | No | No | No | No |
| KO2 | Tgfb3−/− | e13.5 | No | Yes | Yes | No | Perimembranous | No | Yes | No | Yes | Yes |
| KO3 | Tgfb3−/− | e14.5-15.5 | No | No | No | No | No | No | No | No | Yes | Yes |
| KO4 | Tgfb3−/− | e14.5-15.5 | No | Yes | Yes | No | Muscular | No | No | No | Yes | Yes |
| KO5 | Tgfb3−/− | e14.5-15.5 | No | Yes | Yes | No | Muscular | No | Yes | No | No | No |
| KO6 | Tgfb3−/− | e14.5-15.5 | No | No | No | No | No | No | No | No | No | No |
| KO7 | Tgfb3−/− | e14.5-15.5 | No | No | No | No | No | No | No | No | No | No |
| KO8 | Tgfb3−/− | e16.5-17.5 | No | No | No | No | No | No | No | No | No | No |
| KO9 | Tgfb3−/− | e13.5 | No | Yes | Yes | No | Perimembranous | No | No | Yes | Yes | Yes |
| KO10 | Tgfb3−/− | e16.5-17.5 | No | Yes | Yes | No | No | No | No | No | No | No |
| KO11 | Tgfb3−/− | e14.5-15.5 | No | Yes | Yes | ND | No | No | No | Yes | Yes | Yes |
| KO12 | Tgfb3−/− | e18.5 | No | Yes | Yes | Yes | No | No | Yes | No | No | No |
| KO13 | Tgfb3−/− | e18.5 | No | Yes | Yes | Yes | Perimembranous | No | No | Yes | No | No |
| KO14 | Tgfb3−/− | e15.5-16.5 | No | Yes | Yes | No | No | No | Yes | No | No | No |
| KO15 | Tgfb3−/− | e15.5-16.5 | Subaortic | Yes | Yes | Yes | Perimembranous | No | No | Yes | Yes | Yes |
| KO16 | Tgfb3−/− | E18.5 | No | No | No | No | No | No | No | No | No | No |
| KO17 | Tgfb3−/− | e13.5 | No | Yes | Yes | No | No | No | No | No | Yes | Yes |
| KO18 | Tgfb3−/− | e14.5 | No | No | No | No | No | No | No | No | No | No |
| KO19 | Tgfb3−/− | e13.5 | No | Yes | Yes | No | Muscular | No | Yes | No | Yes | Yes |
| Number of cases (%) | 1 (5.2%) | 12 (63.15%) | 12 (63.15%) | 3 (15.7%) | P: 4 (21%); M: 3 (15.7%) | 0 (0%) | 6 (31.5%) | 4 (21%) | 8 (42.1%) | 8 (42.1%) | ||
| Total cases | Tgfb3−/− | 19 | ||||||||||
| Total cases | Wildtype (Tgfb3+/+) | 14 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakrabarti, M.; Al-Sammarraie, N.; Gebere, M.G.; Bhattacharya, A.; Chopra, S.; Johnson, J.; Peña, E.A.; Eberth, J.F.; Poelmann, R.E.; Gittenberger-de Groot, A.C.; et al. Transforming Growth Factor Beta3 is Required for Cardiovascular Development. J. Cardiovasc. Dev. Dis. 2020, 7, 19. https://doi.org/10.3390/jcdd7020019
Chakrabarti M, Al-Sammarraie N, Gebere MG, Bhattacharya A, Chopra S, Johnson J, Peña EA, Eberth JF, Poelmann RE, Gittenberger-de Groot AC, et al. Transforming Growth Factor Beta3 is Required for Cardiovascular Development. Journal of Cardiovascular Development and Disease. 2020; 7(2):19. https://doi.org/10.3390/jcdd7020019
Chicago/Turabian StyleChakrabarti, Mrinmay, Nadia Al-Sammarraie, Mengistu G. Gebere, Aniket Bhattacharya, Sunita Chopra, John Johnson, Edsel A. Peña, John F. Eberth, Robert E. Poelmann, Adriana C. Gittenberger-de Groot, and et al. 2020. "Transforming Growth Factor Beta3 is Required for Cardiovascular Development" Journal of Cardiovascular Development and Disease 7, no. 2: 19. https://doi.org/10.3390/jcdd7020019
APA StyleChakrabarti, M., Al-Sammarraie, N., Gebere, M. G., Bhattacharya, A., Chopra, S., Johnson, J., Peña, E. A., Eberth, J. F., Poelmann, R. E., Gittenberger-de Groot, A. C., & Azhar, M. (2020). Transforming Growth Factor Beta3 is Required for Cardiovascular Development. Journal of Cardiovascular Development and Disease, 7(2), 19. https://doi.org/10.3390/jcdd7020019