
Sox9 Expression in the Second Heart Field; A Morphological Assessment of the Importance to Cardiac Development with Emphasis on Atrioventricular Septation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Histology
2.3. Immunofluorescent Antigen Detection
2.4. AMIRA 3D Analysis
2.5. Cell Profiler Analysis
2.6. Statistical Analysis
3. Results
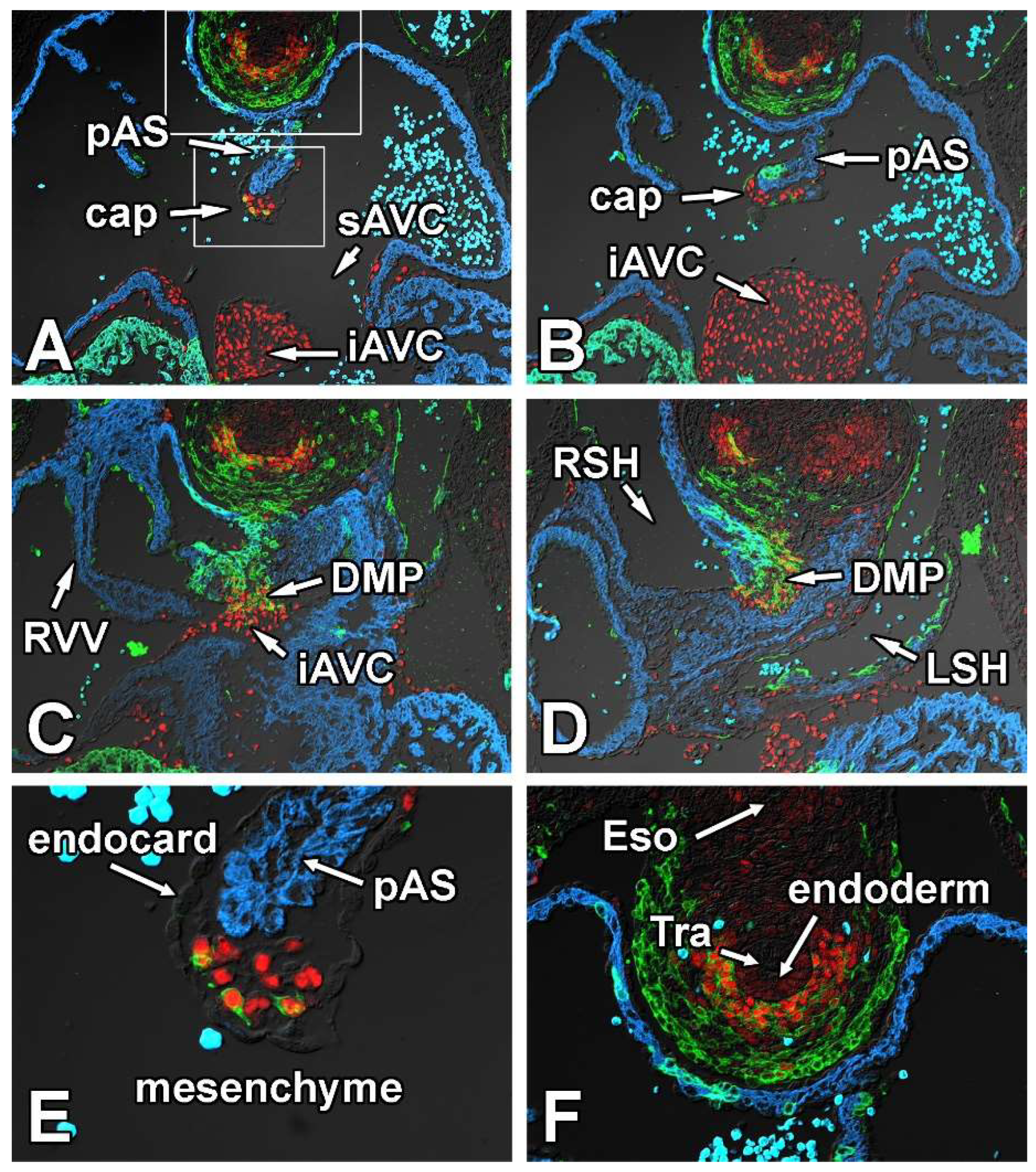
3.1. Sox9 Expression in the Mesenchymal Tissues Contributing to the Atrioventricular Valvuloseptal Complex
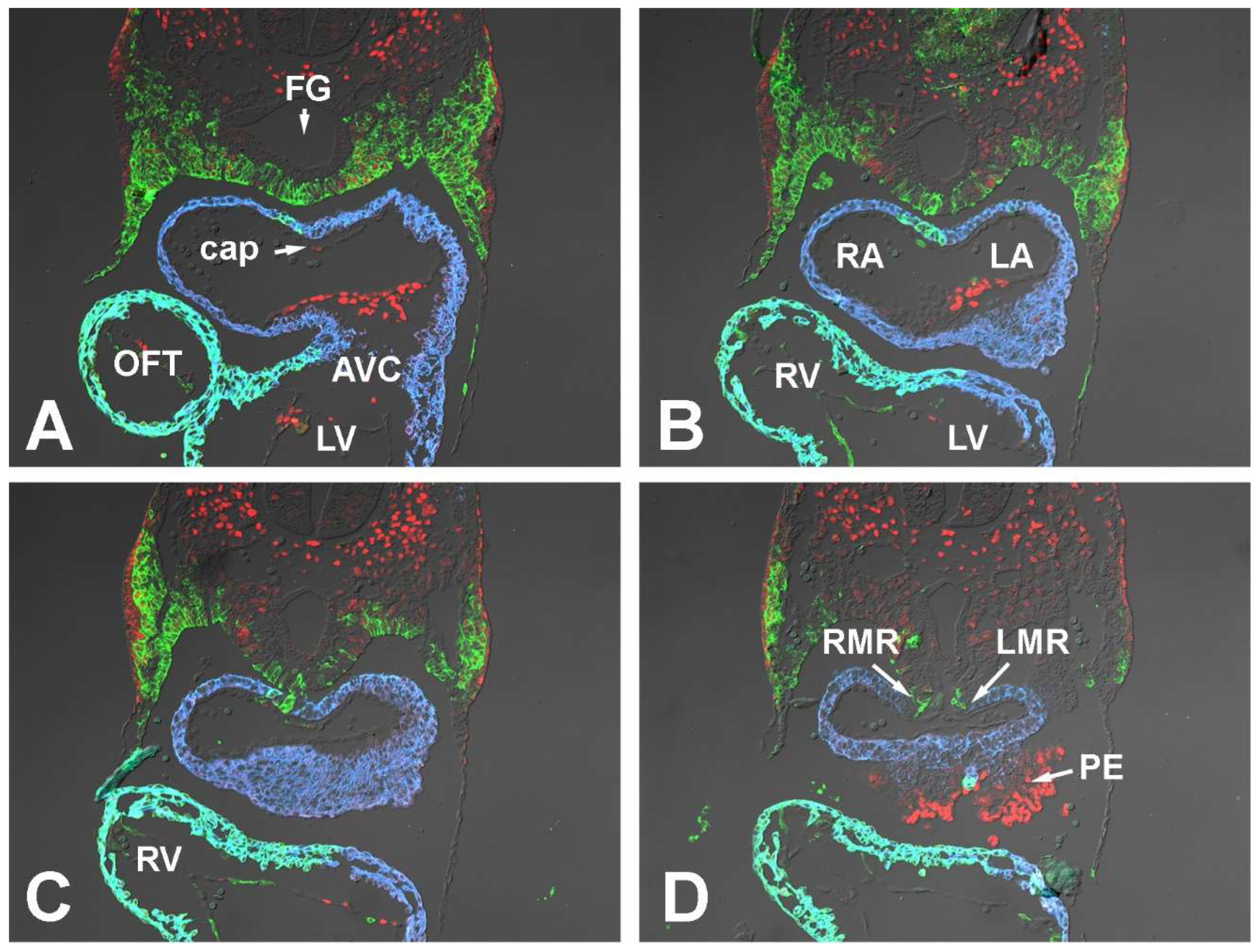
3.1.1. Embryonic Day (ED) 9.5
3.1.2. Embryonic Day 10.5
3.1.3. Embryonic Day 11.5
3.2. Conditional Deletion of Smoothened from the SHF Results in Downregulation of Sox9 in the pSHF
3.3. Creating a SHF-Specific Sox9 Knockout Model to Study Sox9 Dependent Development of the AV Valvuloseptal Complex
3.4. Second Heart Field-Specific Deletion of Sox9 Results in AVSDs and Isolated VSDs
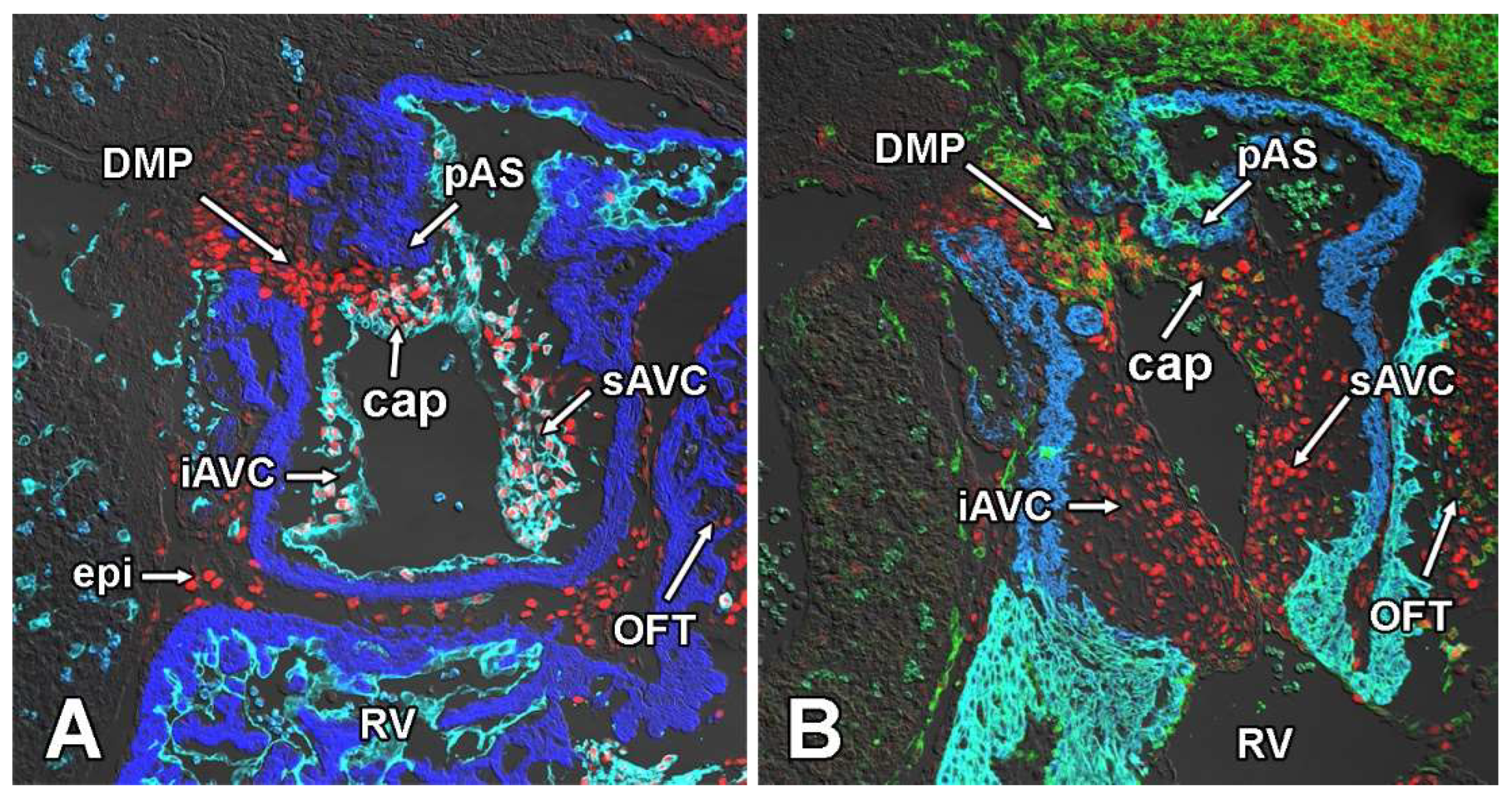
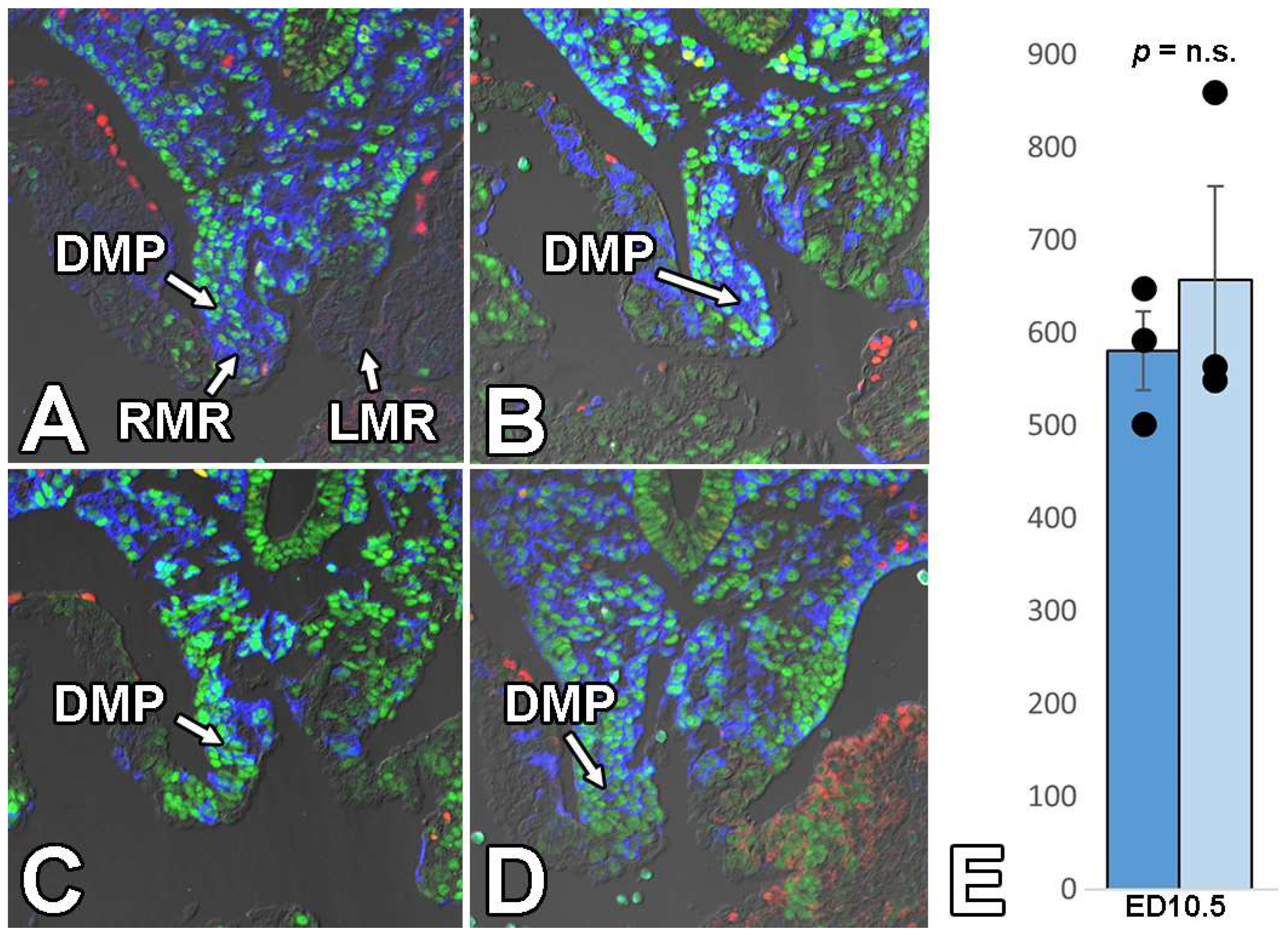
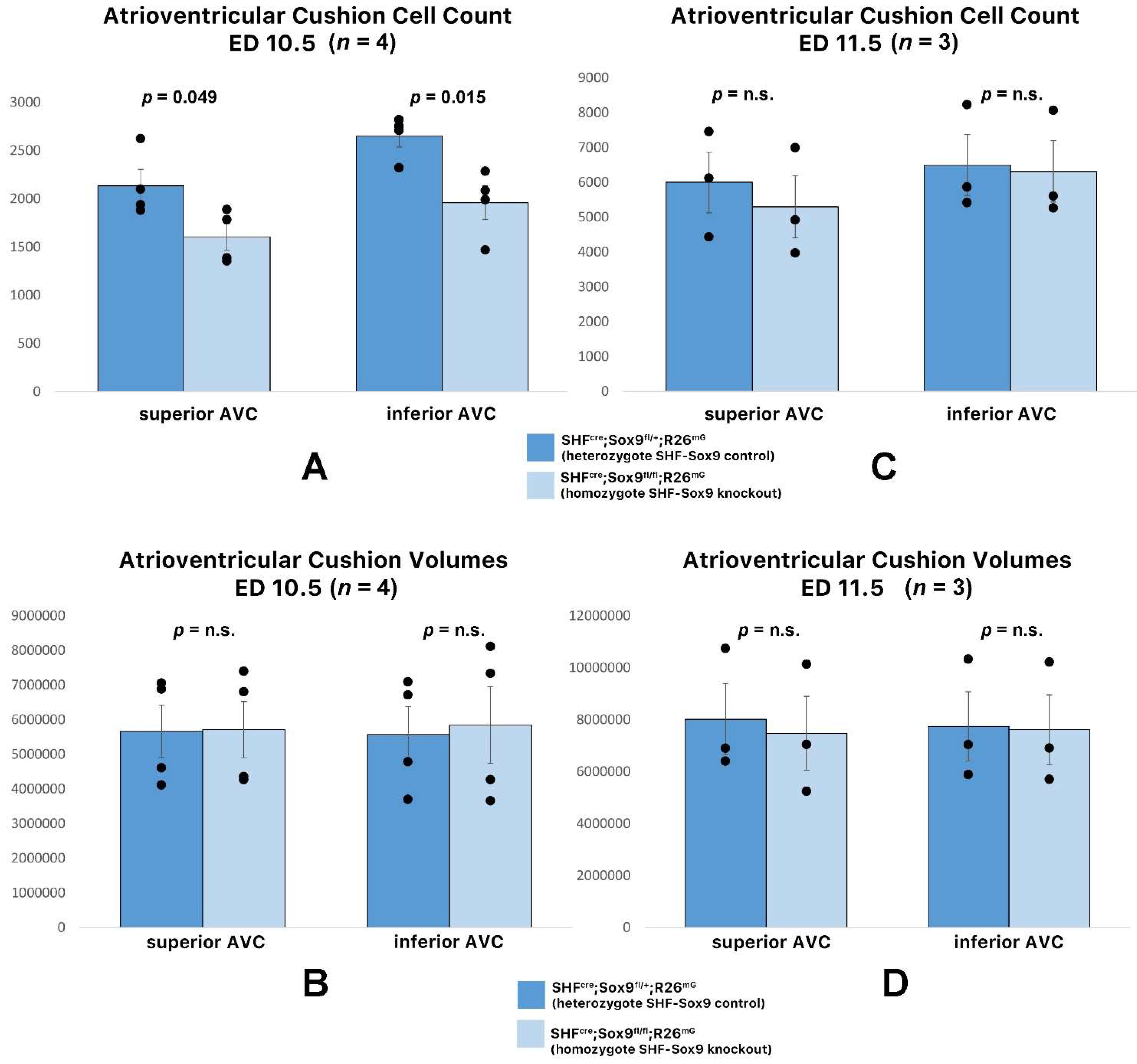
3.5. Deletion of Sox9 from the SHF Does Not Significantly Affect the Initial Development of the DMP and AV Cushions
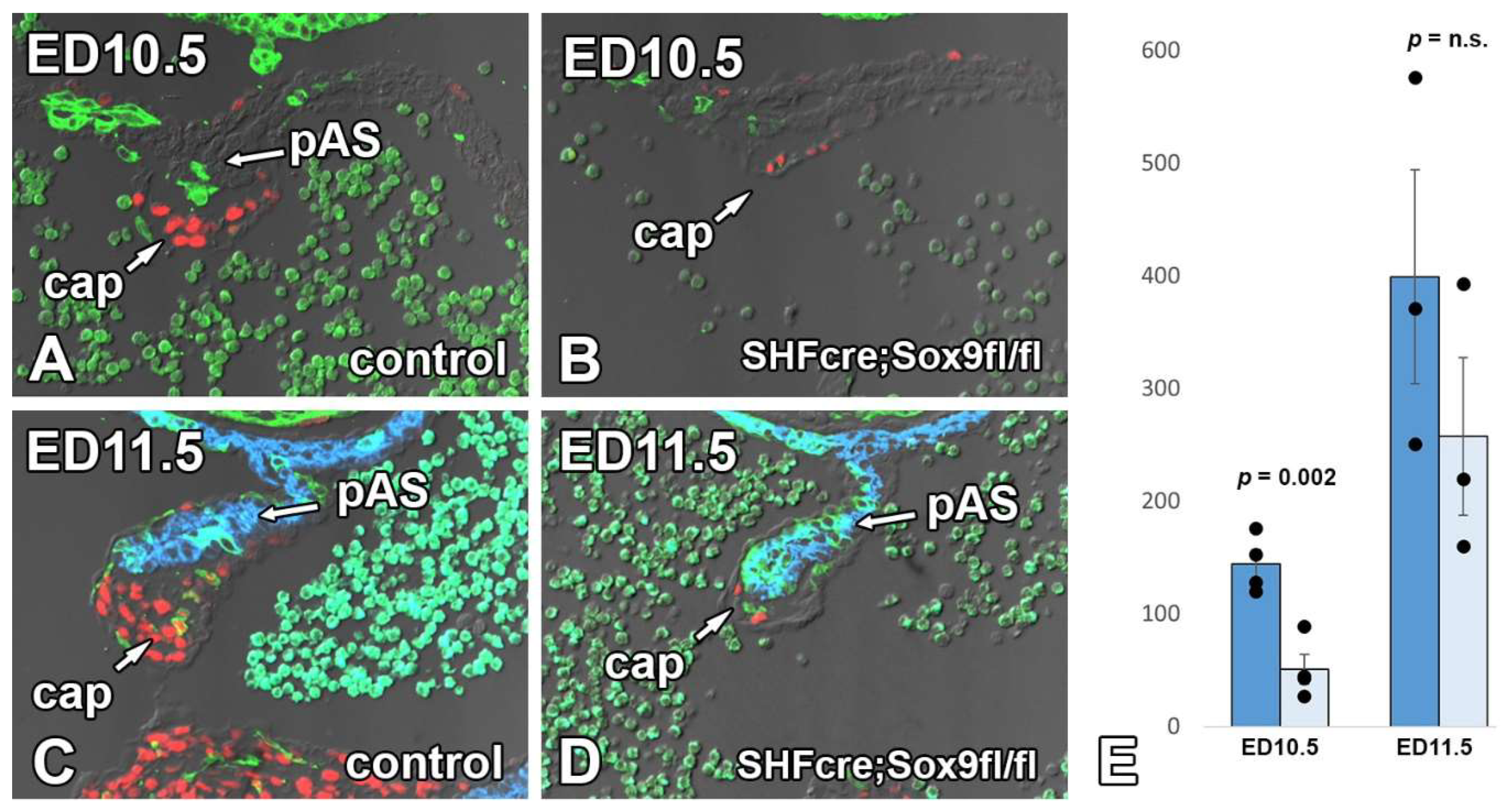
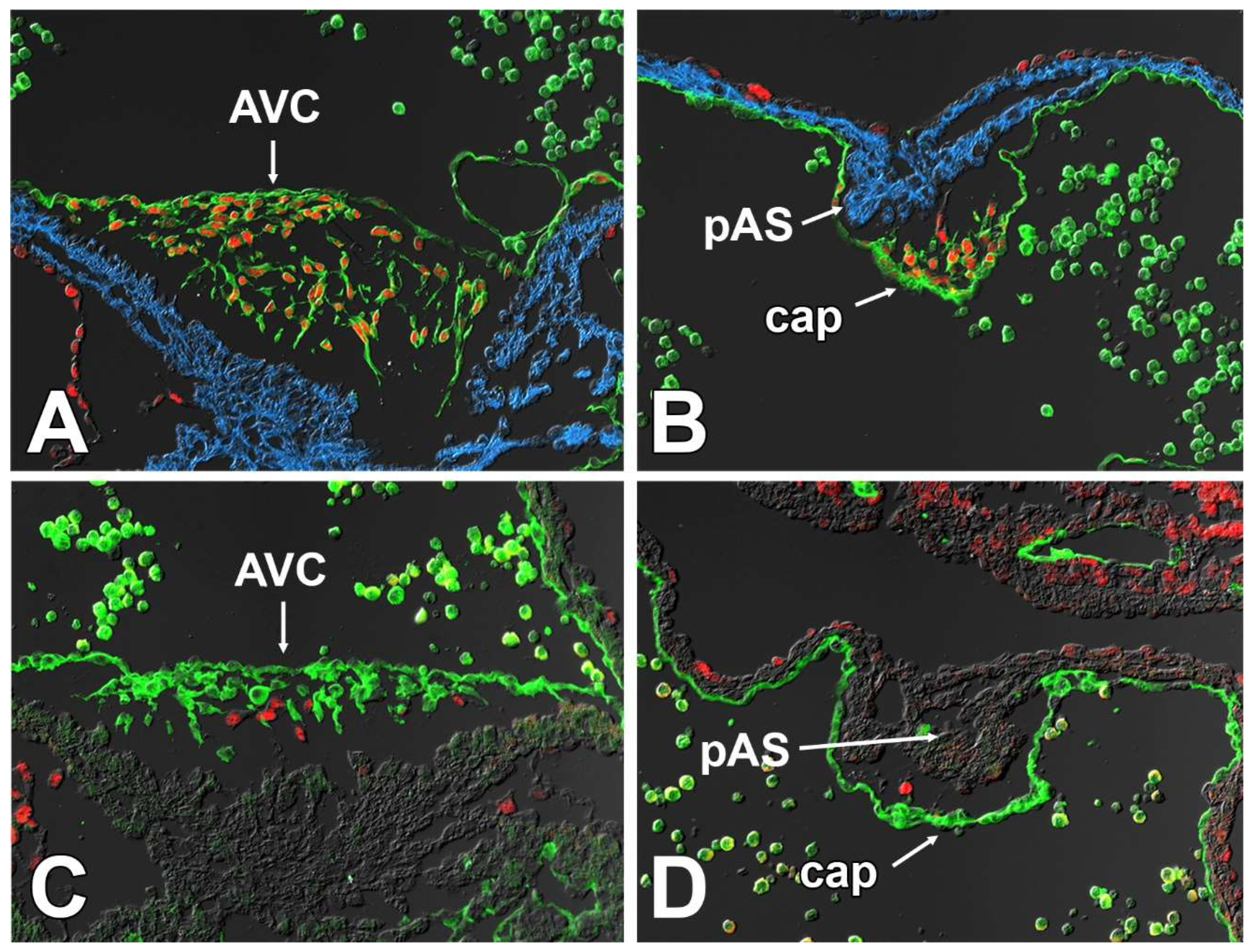
3.6. Deletion of Sox9 from the SHF Leads to Hypoplasia of the Mesenchymal Cap
3.7. Sox9 Expression in the Endocardial Cell Lineage Is Important for the Development of the Mesenchymal Cap
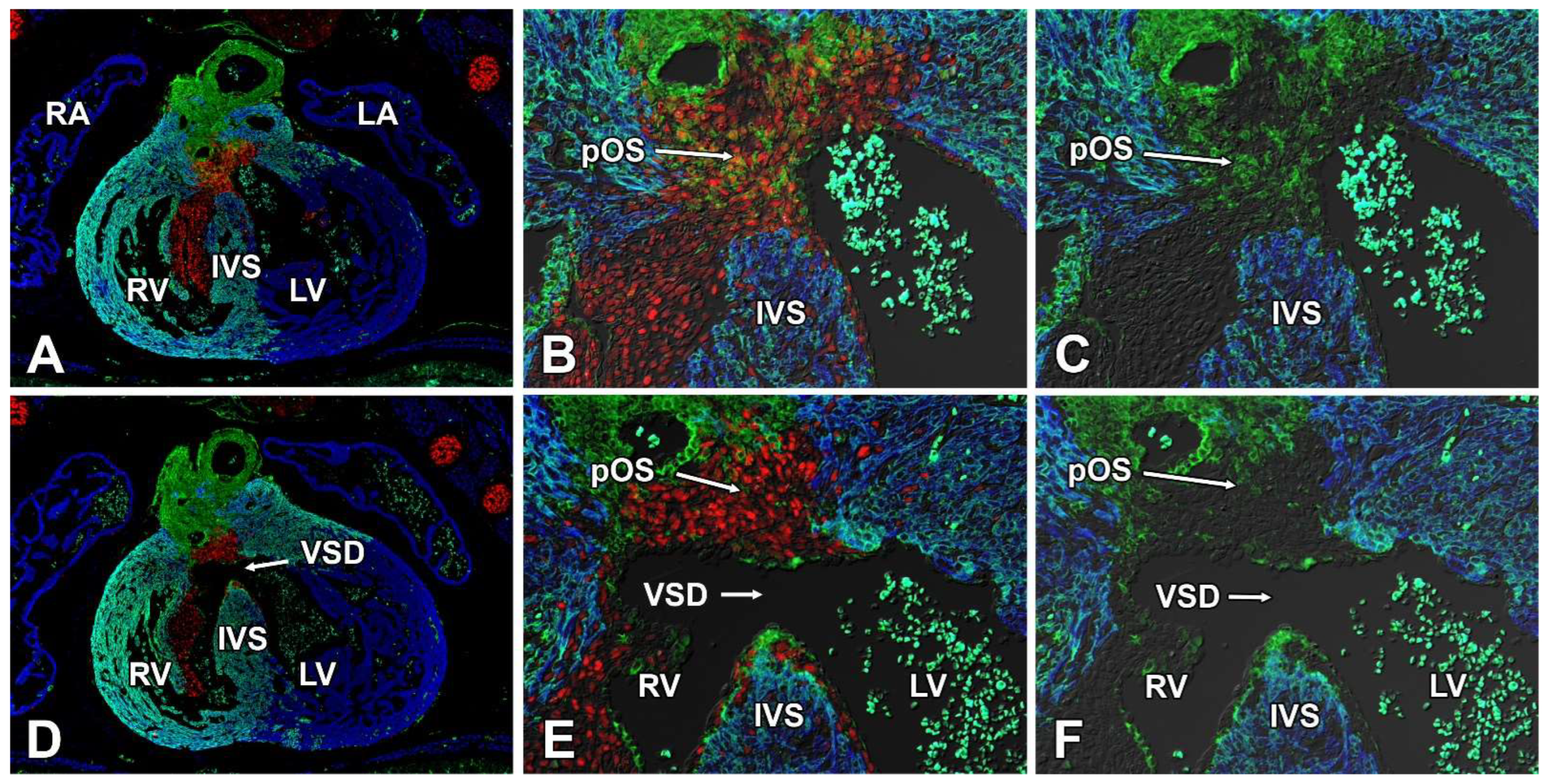
3.8. Deletion of Sox9 from the SHF Causes VSDs
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Snarr, B.S.; O’Neal, J.L.; Chintalapudi, M.R.; Wirrig, E.E.; Phelps, A.L.; Kubalak, S.W.; Wessels, A. Isl1 expression at the venous pole identifies a novel role for the second heart field in cardiac development. Circ. Res. 2007, 101, 971–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, L.E.; Burns, T.A.; Lockhart, M.M.; Phelps, A.L.; Van den Hoff, M.J.; Wessels, A. Wnt/beta-catenin and sonic hedgehog pathways interact in the regulation of the development of the dorsal mesenchymal protrusion. Dev. Dyn. 2016, 245, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, L.E.; Phelps, A.L.; Brown, E.; Kakarla, J.; Anderson, R.H.; van den Hoff, M.J.; Wessels, A. Expression of the bmp receptor alk3 in the second heart field is essential for development of the dorsal mesenchymal protrusion and atrioventricular septation. Circ. Res. 2013, 112, 1420–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goddeeris, M.M.; Rho, S.; Petiet, A.; Davenport, C.L.; Johnson, G.A.; Meyers, E.N.; Klingensmith, J. Intracardiac septation requires hedgehog-dependent cellular contributions from outside the heart. Development 2008, 135, 1887–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calkoen, E.E.; Hazekamp, M.G.; Blom, N.A.; Elders, B.B.; Gittenberger-de Groot, A.C.; Haak, M.C.; Bartelings, M.M.; Roest, A.A.; Jongbloed, M.R. Atrioventricular septal defect: From embryonic development to long-term follow-up. Int. J. Cardiol. 2016, 202, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.I.; Kaplan, S.; Liberthson, R.R. Prevalence of congenital heart disease. Am. Heart J. 2004, 147, 425–439. [Google Scholar] [CrossRef]
- Al-Hay, A.A.; MacNeill, S.J.; Yacoub, M.; Shore, D.F.; Shinebourne, E.A. Complete atrioventricular septal defect, down syndrome, and surgical outcome: Risk factors. Ann. Thorac. Surg. 2003, 75, 412–421. [Google Scholar] [CrossRef]
- Dickinson, D.F.; Arnold, R.; Wilkinson, J.L. Congenital heart disease among 160,480 liveborn children in liverpool 1960 to 1969. Implications for surgical treatment. Br. Heart J. 1981, 46, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Corsten-Janssen, N.; Kerstjens-Frederikse, W.S.; du Marchie Sarvaas, G.J.; Baardman, M.E.; Bakker, M.K.; Bergman, J.E.; Hove, H.D.; Heimdal, K.R.; Rustad, C.F.; Hennekam, R.C.; et al. The cardiac phenotype in patients with a chd7 mutation. Circ. Cardiovasc. Genet. 2013, 6, 248–254. [Google Scholar] [CrossRef] [Green Version]
- Briggs, L.E.; Kakarla, J.; Wessels, A. The pathogenesis of atrial and atrioventricular septal defects with special emphasis on the role of the dorsal mesenchymal protrusion. Differentiation 2012, 84, 117–130. [Google Scholar] [CrossRef]
- Lana-Elola, E.; Watson-Scales, S.; Slender, A.; Gibbins, D.; Martineau, A.; Douglas, C.; Mohun, T.; Fisher, E.M.; Tybulewicz, V. Genetic dissection of down syndrome-associated congenital heart defects using a new mouse mapping panel. eLife 2016, 5, e11614. [Google Scholar] [CrossRef] [Green Version]
- Gittenberger-de Groot, A.C.; Calkoen, E.E.; Poelmann, R.E.; Bartelings, M.M.; Jongbloed, M.R. Morphogenesis and molecular considerations on congenital cardiac septal defects. Ann. Med. 2014, 46, 640–652. [Google Scholar] [CrossRef]
- Snarr, B.S.; Kern, C.B.; Wessels, A. Origin and fate of cardiac mesenchyme. Dev. Dyn. 2008, 237, 2804–2819. [Google Scholar] [CrossRef]
- Snarr, B.S.; Wirrig, E.E.; Phelps, A.L.; Trusk, T.C.; Wessels, A. A spatiotemporal evaluation of the contribution of the dorsal mesenchymal protrusion to cardiac development. Dev. Dyn. 2007, 236, 1287–1294. [Google Scholar] [CrossRef] [Green Version]
- Deepe, R.; Fitzgerald, E.; Wolters, R.; Drummond, J.; Guzman, K.; Hoff, M.; Wessels, A. The mesenchymal cap of the atrial septum and atrial and atrioventricular septation. J. Cardiovasc. Dev. Dis. 2020, 7, 50. [Google Scholar] [CrossRef]
- Campbell, M.; Missen, G.A. Endocardial cushion defects; common atrio-ventricular canal and ostium primum. Br. Heart J. 1957, 19, 403–418. [Google Scholar] [CrossRef] [Green Version]
- McCullough, A. Further examples of endocardial cushion defect in production of a cardiac anomaly complex; a presentation of three cases from autopsy reports. J. Pediatr. 1953, 43, 429–433. [Google Scholar] [CrossRef]
- McCullough, A.W.; Wilbur, E.L. Defect of endocardial cushion development as a source of cardiac anomaly: A presentation of four cases from autopsy reports. Am. J. Pathol. 1944, 20, 321–328. [Google Scholar]
- Van Mierop, L.H.; Alley, R.D. The management of the cleft mitral valve in endocardial cushion defects. Ann. Thorac. Surg. 1966, 2, 416–423. [Google Scholar] [CrossRef]
- Van Mierop, L.H.S.; Alley, R.D.; Kausel, H.W.; Stranahan, A. The anatomy and embryology of endocardial cushion defects. J. Thorac. Cardiov. Surg. 1962, 43, 71. [Google Scholar] [CrossRef]
- Burns, T.; Yang, Y.; Hiriart, E.; Wessels, A. The dorsal mesenchymal protrusion and the pathogenesis of atrioventricular septal defects. J. Cardiovasc. Dev. Dis. 2016, 3, 29. [Google Scholar] [CrossRef] [PubMed]
- Burns, T.A.; Deepe, R.N.; Bullard, J.; Phelps, A.L.; Toomer, K.A.; Hiriart, E.; Norris, R.A.; Haycraft, C.J.; Wessels, A. A novel mouse model for cilia-associated cardiovascular anomalies with a high penetrance of total anomalous pulmonary venous return. Anat. Rec. 2019, 302, 136–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, A.D.; Peterson, M.A.; Friedland-Little, J.M.; Anderson, S.A.; Moskowitz, I.P. Sonic hedgehog is required in pulmonary endoderm for atrial septation. Development 2009, 136, 1761–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Yuan, L.; Goss, A.M.; Wang, T.; Yang, J.; Lepore, J.J.; Zhou, D.; Schwartz, R.J.; Patel, V.; Cohen, E.D.; et al. Characterization and in vivo pharmacological rescue of a wnt2-gata6 pathway required for cardiac inflow tract development. Dev. Cell 2010, 18, 275–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Vliet, P.P.; Lin, L.; Boogerd, C.J.; Martin, J.F.; Andelfinger, G.; Grossfeld, P.D.; Evans, S.M. Tissue specific requirements for wnt11 in developing outflow tract and dorsal mesenchymal protrusion. Dev. Biol. 2017, 429, 249–259. [Google Scholar] [CrossRef]
- Sun, C.; Yu, D.; Ye, W.; Liu, C.; Gu, S.; Sinsheimer, N.R.; Song, Z.; Li, X.; Chen, C.; Song, Y.; et al. The short stature homeobox 2 (shox2)-bone morphogenetic protein (bmp) pathway regulates dorsal mesenchymal protrusion development and its temporary function as a pacemaker during cardiogenesis. J. Biol. Chem. 2015, 290, 2007–2023. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Hoffmann, A.D.; Burnicka-Turek, O.; Friedland-Little, J.M.; Zhang, K.; Moskowitz, I.P. Tbx5-hedgehog molecular networks are essential in the second heart field for atrial septation. Dev. Cell 2012, 23, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Gallina, D.; Lincoln, J. Dynamic expression profiles of sox9 in embryonic, post natal, and adult heart valve cell populations. Anat. Rec. 2018, 302, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Lincoln, J.; Kist, R.; Scherer, G.; Yutzey, K.E. Sox9 is required for precursor cell expansion and extracellular matrix organization during mouse heart valve development. Dev. Biol. 2007, 305, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; Chaboissier, M.C.; Behringer, R.R.; Rowitch, D.H.; Schedl, A.; Epstein, J.A.; de Crombrugghe, B. Essential role of sox9 in the pathway that controls formation of cardiac valves and septa. Proc. Natl. Acad. Sci. USA 2004, 101, 6502–6507. [Google Scholar] [CrossRef] [Green Version]
- Garside, V.C.; Cullum, R.; Alder, O.; Lu, D.Y.; Vander Werff, R.; Bilenky, M.; Zhao, Y.; Jones, S.J.; Marra, M.A.; Underhill, T.M.; et al. Sox9 modulates the expression of key transcription factors required for heart valve development. Development 2015, 142, 4340–4350. [Google Scholar] [CrossRef]
- Verzi, M.P.; McCulley, D.J.; De Val, S.; Dodou, E.; Black, B.L. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 2005, 287, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Kisanuki, Y.Y.; Hammer, R.E.; Miyazaki, J.; Williams, S.C.; Richardson, J.A.; Yanagisawa, M. Tie2-cre transgenic mice: A new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 2001, 230, 230–242. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Zhang, Z.; Lui, W.; Chen, X.; Wang, Y.; Chamberlain, A.A.; Moreno-Rodriguez, R.A.; Markwald, R.R.; O’Rourke, B.P.; Sharp, D.J.; et al. Endocardial cells form the coronary arteries by angiogenesis through myocardial-endocardial vegf signaling. Cell 2012, 151, 1083–1096. [Google Scholar] [CrossRef] [Green Version]
- Waller, B.R., 3rd; Wessels, A. Cardiac morphogenesis and dysmorphogenesis. An immunohistochemical approach. Methods Mol. Biol. 2000, 135, 151–161. [Google Scholar]
- Lockhart, M.M.; Boukens, B.J.; Phelps, A.; Brown, C.-L.M.; Toomer, K.A.; Burns, T.A.; Mukherjee, R.D.; Norris, R.A.; Trusk, T.C.; van den Hoff, M.J.B.; et al. Alk3 mediated bmp signaling controls the contribution of epicardially derived cells to the tissues of the atrioventricular junction. Dev. Biol. 2014, 396, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Mommersteeg, M.T.; Soufan, A.T.; de Lange, F.J.; van den Hoff, M.J.; Anderson, R.H.; Christoffels, V.M.; Moorman, A.F. Two distinct pools of mesenchyme contribute to the development of the atrial septum. Circ. Res. 2006, 99, 351–353. [Google Scholar] [CrossRef]
- Smith, C.; Baek, S.; Sung, C.; Tallquist, M. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require pdgf receptor signaling. Circ. Res. 2011, 108, 26. [Google Scholar] [CrossRef]
- Wessels, A.; van den Hoff, M.J.; Adamo, R.F.; Phelps, A.L.; Lockhart, M.M.; Sauls, K.; Briggs, L.E.; Norris, R.A.; van Wijk, B.; Perez-Pomares, J.M.; et al. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 2012, 366, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Zhang, J.J.; Moro, A.; Kushida, M.; Wegner, M.; Kim, P.C. Regulation of sox9 by sonic hedgehog (shh) is essential for patterning and formation of tracheal cartilage. Dev. Dyn. 2010, 239, 514–526. [Google Scholar] [CrossRef]
- Li, X.M.; Piao, Y.J.; Sohn, K.C.; Ha, J.M.; Im, M.; Seo, Y.J.; Whang, K.U.; Lee, J.H.; Lee, Y.; Kim, C.D. Sox9 is a beta-catenin-regulated transcription factor that enhances the colony-forming activity of squamous cell carcinoma cells. Mol. Med. Rep. 2016, 14, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodosiou, N.A.; Tabin, C.J. Sox9 and nkx2.5 determine the pyloric sphincter epithelium under the control of bmp signaling. Dev. Biol. 2005, 279, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Blache, P.; van de Wetering, M.; Duluc, I.; Domon, C.; Berta, P.; Freund, J.N.; Clevers, H.; Jay, P. Sox9 is an intestine crypt transcription factor, is regulated by the wnt pathway, and represses the cdx2 and muc2 genes. J. Cell Biol. 2004, 166, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchett, J.; Harvey, E.; Athwal, V.; Berry, A.; Rowe, C.; Oakley, F.; Moles, A.; Mann, D.A.; Bobola, N.; Sharrocks, A.D.; et al. Osteopontin is a novel downstream target of sox9 with diagnostic implications for progression of liver fibrosis in humans. Hepatology 2012, 56, 1108–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Es, J.H.; Jay, P.; Gregorieff, A.; van Gijn, M.E.; Jonkheer, S.; Hatzis, P.; Thiele, A.; van den Born, M.; Begthel, H.; Brabletz, T.; et al. Wnt signalling induces maturation of paneth cells in intestinal crypts. Nat. Cell Biol. 2005, 7, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Dyer, L.A.; Kirby, M.L. Sonic hedgehog maintains proliferation in secondary heart field progenitors and is required for normal arterial pole formation. Dev. Biol. 2009, 330, 305–317. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, A.; Samtani, R.; Dhanantwari, P.; Lee, E.; Yamada, S.; Shiota, K.; Donofrio, M.T.; Leatherbury, L.; Lo, C.W. A detailed comparison of mouse and human cardiac development. Pediatr. Res. 2014, 76, 500–507. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.H.; Spicer, D.E.; Mohun, T.J.; Hikspoors, J.; Lamers, W.H. Remodeling of the embryonic interventricular communication in regard to the description and classification of ventricular septal defects. Anat. Rec. 2019, 302, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Houston, C.S.; Opitz, J.M.; Spranger, J.W.; Macpherson, R.I.; Reed, M.H.; Gilbert, E.F.; Herrmann, J.; Schinzel, A. The campomelic syndrome: Review, report of 17 cases, and follow-up on the currently 17-year-old boy first reported by maroteaux et al. in 1971. Am. J. Med. Genet. 1983, 15, 3–28. [Google Scholar] [CrossRef]
- Mansour, S.; Hall, C.M.; Pembrey, M.E.; Young, I.D. A clinical and genetic study of campomelic dysplasia. J. Med. Genet. 1995, 32, 415–420. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Castro, M.; Gordon, C.T.; Petit, F.; Nord, A.S.; Callier, P.; Andrieux, J.; Guerin, P.; Pichon, O.; David, A.; Abadie, V.; et al. Congenital heart defects in patients with deletions upstream of sox9. Hum. Mutat. 2013, 34, 1628–1631. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deepe, R.N.; Drummond, J.R.; Wolters, R.A.; Fitzgerald, E.A.; Tarolli, H.G.; Harvey, A.B.; Wessels, A. Sox9 Expression in the Second Heart Field; A Morphological Assessment of the Importance to Cardiac Development with Emphasis on Atrioventricular Septation. J. Cardiovasc. Dev. Dis. 2022, 9, 376. https://doi.org/10.3390/jcdd9110376
Deepe RN, Drummond JR, Wolters RA, Fitzgerald EA, Tarolli HG, Harvey AB, Wessels A. Sox9 Expression in the Second Heart Field; A Morphological Assessment of the Importance to Cardiac Development with Emphasis on Atrioventricular Septation. Journal of Cardiovascular Development and Disease. 2022; 9(11):376. https://doi.org/10.3390/jcdd9110376
Chicago/Turabian StyleDeepe, Raymond N., Jenna R. Drummond, Renélyn A. Wolters, Emily A. Fitzgerald, Hannah G. Tarolli, Andrew B. Harvey, and Andy Wessels. 2022. "Sox9 Expression in the Second Heart Field; A Morphological Assessment of the Importance to Cardiac Development with Emphasis on Atrioventricular Septation" Journal of Cardiovascular Development and Disease 9, no. 11: 376. https://doi.org/10.3390/jcdd9110376