
Lipopolysaccharide Modifies Sodium Current Kinetics through ROS and PKC Signalling in Induced Pluripotent Stem-Derived Cardiomyocytes from Brugada Syndrome Patient

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Generation of Human iPS Cells
2.3. Generation of hiPSC-CMs
2.4. ROS Detection
2.5. Patch-Clamp
2.6. Drugs
2.7. Statistics
3. Results
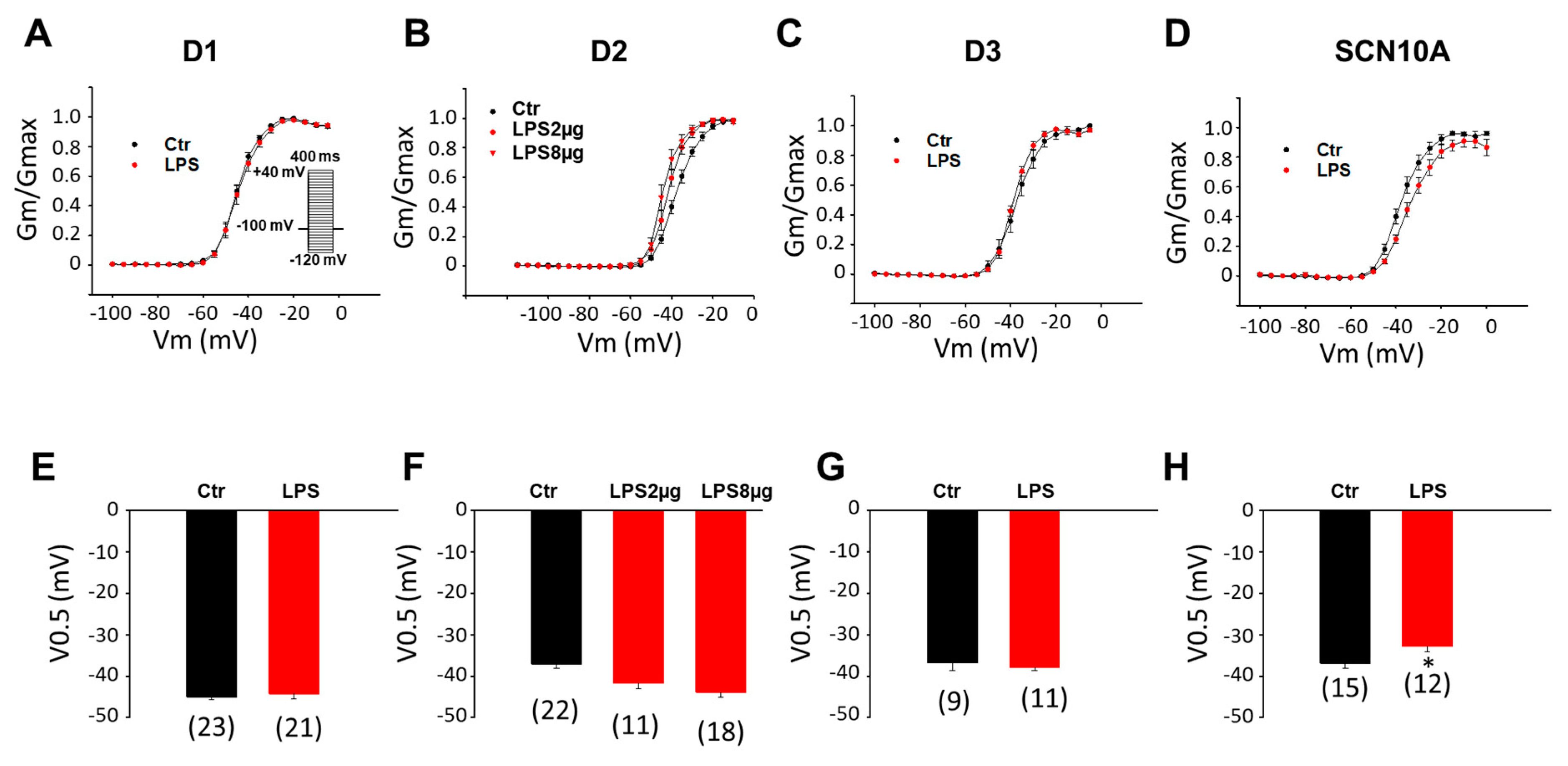
3.1. LPS Reduced Peak INa in hiPSC-CMs Derived from BrS-Patients
3.2. ROS and PKC Blocker Attenuated Effects of LPS on Peak INa
3.3. Peroxide Decreased the Peak INa in BrS-hiPSC-CMs
H2O2 Effect Was Blocked by a PKC Inhibitor
4. Discussion
5. Conclusions
6. Study Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brugada, J.; Campuzano, O.; Arbelo, E.; Sarquella-Brugada, G.; Brugada, R. Present Status of Brugada Syndrome: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- Hoogendijk, M.G.; Potse, M.; Vinet, A.; de Bakker, J.M.; Coronel, R. ST segment elevation by current-to-load mismatch: An experimental and computational study. Heart Rhythm 2011, 8, 111–118. [Google Scholar] [CrossRef]
- Vohra, J.; Rajagopalan, S. Update on the Diagnosis and Management of Brugada Syndrome. Heart Lung Circ. 2015, 24, 1141–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Kirsch, G.E.; Zhang, D.; Brugada, R.; Brugada, J.; Brugada, P.; Potenza, D.; Moya, A.; Borggrefe, M.; Breithardt, G. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998, 392, 293. [Google Scholar] [CrossRef]
- Campuzano, O.; Sarquella-Brugada, G.; Fernandez-Falgueras, A.; Cesar, S.; Coll, M.; Mates, J.; Arbelo, E.; Perez-Serra, A.; Del Olmo, B.; Jordá, P.; et al. Genetic interpretation and clinical translation of minor genes related to Brugada syndrome. Hum. Mutat. 2019, 40, 749–764. [Google Scholar] [CrossRef]
- Hosseini, S.M.; Kim, R.; Udupa, S.; Costain, G.; Jobling, R.; Liston, E.; Jamal, S.M.; Szybowska, M.; Morel, C.F.; Bowdin, S. Reappraisal of reported genes for sudden arrhythmic death: Evidence-based evaluation of gene validity for Brugada syndrome. Circulation 2018, 138, 1195–1205. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Albers, S.; Cyganek, L.; Zhao, Z.; Lan, H.; Li, X.; Xu, Q.; Kleinsorge, M.; Huang, M.; Liao, Z.; et al. A cellular model of Brugada syndrome with SCN10A variants using human-induced pluripotent stem cell-derived cardiomyocytes. Europace 2019, 21, 1410–1421. [Google Scholar] [CrossRef]
- Yu, G.; Liu, Y.; Qin, J.; Wang, Z.; Hu, Y.; Wang, F.; Li, Y.; Chakrabarti, S.; Chen, Q.; Wang, Q.K. Mechanistic insights into the interaction of the MOG1 protein with the cardiac sodium channel Nav1. 5 clarify the molecular basis of Brugada syndrome. J. Biol. Chem. 2018, 293, 18207–18217. [Google Scholar] [CrossRef] [Green Version]
- Campuzano, O.; Berne, P.; Selga, E.; Allegue, C.; Iglesias, A.; Brugada, J.; Brugada, R. Brugada syndrome and p.E61X_RANGRF. Cardiol. J. 2014, 21, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Chen, Y.Q.; Fan, L.L.; Guo, S.; Li, J.J.; Jin, J.Y.; Xiang, R. Whole-exome sequencing identifies a novel mutation of GPD1L (R189X) associated with familial conduction disease and sudden death. J. Cell. Mol. Med. 2018, 22, 1350–1354. [Google Scholar] [CrossRef]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campuzano, O.; Fernandez-Falgueras, A.; Lemus, X.; Sarquella-Brugada, G.; Cesar, S.; Coll, M.; Mates, J.; Arbelo, E.; Jorda, P.; Perez-Serra, A.; et al. Short QT Syndrome: A Comprehensive Genetic Interpretation and Clinical Translation of Rare Variants. J. Clin. Med. 2019, 8, 1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, T.; Wu, J.; Kaneko, Y.; Ashihara, T.; Ohno, S.; Irie, T.; Ding, W.G.; Matsuura, H.; Kurabayashi, M.; Horie, M. KCNE3 T4A as the Genetic Basis of Brugada-Pattern Electrocardiogram. Circ. J. 2012, 76, 2763–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, S.; Zankov, D.P.; Ding, W.G.; Itoh, H.; Makiyama, T.; Doi, T.; Shizuta, S.; Hattori, T.; Miyamoto, A.; Naiki, N.; et al. KCNE5 (KCNE1L) Variants Are Novel Modulators of Brugada Syndrome and Idiopathic Ventricular Fibrillation. Circ. Arrhythm. Electrophysiol. 2011, 4, 352–361. [Google Scholar] [CrossRef] [Green Version]
- Cerrone, M.; Priori, S.G. Genetics of sudden death: Focus on inherited channelopathies. Eur. Heart J. 2011, 32, 2109–2118. [Google Scholar] [CrossRef] [Green Version]
- Lazzerini, P.E.; Capecchi, P.L.; Laghi-Pasini, F.; Boutjdir, M. Autoimmune channelopathies as a novel mechanism in cardiac arrhythmias. Nat. Rev. Cardiol. 2017, 14, 521. [Google Scholar] [CrossRef]
- Capecchi, P.L.; Laghi-Pasini, F.; El-Sherif, N.; Qu, Y.; Boutjdir, M.; Lazzerini, P.E. Autoimmune and inflammatory K(+) channelopathies in cardiac arrhythmias: Clinical evidence and molecular mechanisms. Heart Rhythm 2019, 16, 1273–1280. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, Q.; Zeng, Z.; Li, Q.; Zhou, S.; Zhou, M.; Xue, Y.; Cheng, X.; Xia, Y.; Wang, Q.; et al. Regulation of SCN3B/scn3b by Interleukin 2 (IL-2): IL-2 modulates SCN3B/scn3b transcript expression and increases sodium current in myocardial cells. BMC Cardiovasc. Disord. 2016, 16, 1. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Qin, L.; Li, J. Enhancement by TNF-α of TTX-resistant Na(V) current in muscle sensory neurons after femoral artery occlusion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R772–R780. [Google Scholar] [CrossRef]
- Casado-Arroyo, R.; Berne, P.; Rao, J.Y.; Rodriguez-Manero, M.; Levinstein, M.; Conte, G.; Sieira, J.; Namdar, M.; Ricciardi, D.; Chierchia, G.B.; et al. Long-Term Trends in Newly Diagnosed Brugada Syndrome: Implications for Risk Stratification. J. Am. Coll. Cardiol. 2016, 68, 614–623. [Google Scholar] [CrossRef]
- Frustaci, A.; Priori, S.G.; Pieroni, M.; Chimenti, C.; Napolitano, C.; Rivolta, I.; Sanna, T.; Bellocci, F.; Russo, M.A. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation 2005, 112, 3680–3687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonny, A.; Tonet, J.; Marquez, M.F.; De Sisti, A.; Temfemo, A.; Himbert, C.; Gueffaf, F.; Larrazet, F.; Ditah, I.; Frank, R.; et al. C-reactive protein levels in the brugada syndrome. Cardiol. Res. Pract. 2011, 2011, 341521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Tung, R.; Shivkumar, K.; Bradfield, J.S. Brugada syndrome-Malignant phenotype associated with acute cardiac inflammation? HeartRhythm Case Rep. 2017, 3, 384–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouda, M.A.; Ruben, P.C. Protein Kinases Mediate Anti-Inflammatory Effects of Cannabidiol and Estradiol against High Glucose in Cardiac Sodium Channels. Front. Pharmacol. 2021, 12, 668657. [Google Scholar] [CrossRef] [PubMed]
- El-Battrawy, I.; Lan, H.; Cyganek, L.; Zhao, Z.; Li, X.; Buljubasic, F.; Lang, S.; Yucel, G.; Sattler, K.; Zimmermann, W.H.; et al. Modeling Short QT Syndrome Using Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. J. Am. Heart Assoc. 2018, 7, e007394. [Google Scholar] [CrossRef] [Green Version]
- Sattler, K.; El-Battrawy, I.; Cyganek, L.; Lang, S.; Lan, H.; Li, X.; Zhao, Z.; Utikal, J.; Wieland, T.; Borggrefe, M.; et al. TRPV1 activation and internalization is part of the LPS-induced inflammation in human iPSC-derived cardiomyocytes. Sci. Rep. 2021, 11, 14689. [Google Scholar] [CrossRef]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857. [Google Scholar] [CrossRef] [Green Version]
- Yücel, G.; Zhao, Z.; El-Battrawy, I.; Lan, H.; Lang, S.; Li, X.; Buljubasic, F.; Zimmermann, W.-H.; Cyganek, L.; Utikal, J.; et al. Lipopolysaccharides induced inflammatory responses and electrophysiological dysfunctions in human-induced pluripotent stem cell derived cardiomyocytes. Sci. Rep. 2017, 7, 2935. [Google Scholar] [CrossRef]
- Wang, H.J.; Li, Y.L.; Zhang, L.B.; Zucker, I.H.; Gao, L.; Zimmerman, M.C.; Wang, W. Endogenous reactive oxygen species modulates voltage-gated sodium channels in dorsal root ganglia of rats. J. Appl. Physiol. 2011, 110, 1439–1447. [Google Scholar] [CrossRef]
- Liu, M.; Liu, H.; Dudley, S.C., Jr. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ. Res. 2010, 107, 967–974. [Google Scholar] [CrossRef] [Green Version]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxidative Med. Cell. Longev. 2016, 2016, 7432797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, L.C.; Kokkinaki, D.; Valenti, M.C.; Kim, G.J.; Barca, E.; Tomar, D.; Hoffman, N.E.; Subramanyam, P.; Colecraft, H.M.; Hirano, M.; et al. Inhibition of NADPH oxidase 2 (NOX2) prevents sepsis-induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI Insight 2017, 2, e94248. [Google Scholar] [CrossRef]
- Dikalov, S.; Griendling, K.K.; Harrison, D.G. Measurement of reactive oxygen species in cardiovascular studies. Hypertension 2007, 49, 717–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonderlin, N.; Siebermair, J.; Kaya, E.; Köhler, M.; Rassaf, T.; Wakili, R. Critical inflammatory mechanisms underlying arrhythmias. Herz 2019, 44, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Bartekova, M.; Radosinska, J.; Jelemensky, M.; Dhalla, N.S. Role of cytokines and inflammation in heart function during health and disease. Heart Fail. Rev. 2018, 23, 733–758. [Google Scholar] [CrossRef]
- Lee, S.-H.; Chen, Y.-C.; Chen, Y.-J.; Chang, S.-L.; Tai, C.-T.; Wongcharoen, W.; Yeh, H.-I.; Lin, C.-I.; Chen, S.-A. Tumor necrosis factor-α alters calcium handling and increases arrhythmogenesis of pulmonary vein cardiomyocytes. Life Sci. 2007, 80, 1806–1815. [Google Scholar] [CrossRef]
- Lu, Y.; Peng, F.; Dong, M.; Yang, H. Endocannabinoid 2-arachidonylglycerol protects primary cultured neurons against LPS-induced impairments in rat caudate nucleus. J. Mol. Neurosci. 2014, 54, 49–58. [Google Scholar] [CrossRef]
- Hwang, H.R.; Tai, B.Y.; Cheng, P.Y.; Chen, P.N.; Sung, P.J.; Wen, Z.H.; Hsu, C.H. Excavatolide B Modulates the Electrophysiological Characteristics and Calcium Homeostasis of Atrial Myocytes. Mar. Drugs 2017, 15, 25. [Google Scholar] [CrossRef] [Green Version]
- Tai, B.Y.; Wen, Z.H.; Cheng, P.Y.; Yang, H.Y.; Duh, C.Y.; Chen, P.N.; Hsu, C.H. Lemnalol Modulates the Electrophysiological Characteristics and Calcium Homeostasis of Atrial Myocytes. Mar. Drugs 2019, 17, 619. [Google Scholar] [CrossRef] [Green Version]
- Koesters, A.; Engisch, K.L.; Rich, M.M. Decreased cardiac excitability secondary to reduction of sodium current may be a significant contributor to reduced contractility in a rat model of sepsis. Crit. Care 2014, 18, R54. [Google Scholar] [CrossRef] [Green Version]
- Shang, F.; Zhao, L.; Zheng, Q.; Wang, J.; Xu, Z.; Liang, W.; Liu, H.; Liu, S.; Zhang, L. Simvastatin inhibits lipopolysaccharide-induced tumor necrosis factor-alpha expression in neonatal rat cardiomyocytes: The role of reactive oxygen species. Biochem. Biophys. Res. Commun. 2006, 351, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Peng, W.; Zheng, Y.; Hao, H.; Li, S.; Yao, Y.; Ding, Y.; Zhang, J.; Lyu, J.; Zeng, Q. Upregulation of UCP2 Expression Protects against LPS-Induced Oxidative Stress and Apoptosis in Cardiomyocytes. Oxidative Med. Cell. Longev. 2019, 2019, 2758262. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Nakazato, Y. Brugada-like electrocardiogram detected after reconstructive operation for oesophageal cancer. Europace 2010, 12, 1542. [Google Scholar] [CrossRef] [PubMed]
- Tarín, N.; Farré, J.; Rubio, J.M.; Tuñón, J.; Castro-Dorticós, J. Brugada-like electrocardiographic pattern in a patient with a mediastinal tumor. Pacing Clin. Electrophysiol. PACE 1999, 22, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Kusano, K.F. Brugada phenotype and prostate cancer. Circ. J. Off. J. Jpn. Circ. Soc. 2009, 73, 35–36. [Google Scholar] [CrossRef] [Green Version]
- Haseeb, S.; Kariyanna, P.T.; Jayarangaiah, A.; Thirunavukkarasu, G.; Hegde, S.; Marmur, J.D.; Neurgaonkar, S.; McFarlane, S.I. Brugada Pattern in Diabetic Ketoacidosis: A Case Report and Scoping Study. Am. J. Med. Case Rep. 2018, 6, 173–179. [Google Scholar] [CrossRef]
- Anselm, D.D.; Gottschalk, B.H.; Baranchuk, A. Brugada phenocopies: Consideration of morphologic criteria and early findings from an international registry. Can. J. Cardiol. 2014, 30, 1511–1515. [Google Scholar] [CrossRef]
- Batra, A.S.; Watson, R.; McCanta, A.C. Exercise-induced syncope and Brugada syndrome. Ann. Pediatr. Cardiol. 2019, 12, 292–294. [Google Scholar] [CrossRef]
- Wu, C.I.; Postema, P.G.; Arbelo, E.; Behr, E.R.; Bezzina, C.R.; Napolitano, C.; Robyns, T.; Probst, V.; Schulze-Bahr, E.; Remme, C.A.; et al. SARS-CoV-2, COVID-19, and inherited arrhythmia syndromes. Heart Rhythm 2020, 17, 1456–1462. [Google Scholar] [CrossRef]
- Joseph, L.C.; Reyes, M.V.; Lakkadi, K.R.; Gowen, B.H.; Hasko, G.; Drosatos, K.; Morrow, J.P. PKCδ causes sepsis-induced cardiomyopathy by inducing mitochondrial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H778–H786. [Google Scholar] [CrossRef]
- Herbert, J.; Augereau, J.; Gleye, J.; Maffrand, J. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun. 1990, 172, 993–999. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A.; Kang, D. The role of peroxiredoxins in the transduction of H2O2 signals. Antioxid. Redox Signal. 2018, 28, 537–557. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.A.; Giles, W.R. Ionic mechanism of the effects of hydrogen peroxide in rat ventricular myocytes. J. Physiol. 1997, 500 Pt 3, 631–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.; Rogers, J.C.; Tanada, T.N.; Catterall, W.A.; Scheuer, T. Phosphorylation of S1505 in the cardiac Na+ channel inactivation gate is required for modulation by protein kinase C. J. Gen. Physiol. 1996, 108, 375–379. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, Z.; Li, Y.; Fan, X.; Yang, Z.; El-Battrawy, I.; Zhou, X.; Akin, I. Lipopolysaccharide Modifies Sodium Current Kinetics through ROS and PKC Signalling in Induced Pluripotent Stem-Derived Cardiomyocytes from Brugada Syndrome Patient. J. Cardiovasc. Dev. Dis. 2022, 9, 119. https://doi.org/10.3390/jcdd9040119
Liao Z, Li Y, Fan X, Yang Z, El-Battrawy I, Zhou X, Akin I. Lipopolysaccharide Modifies Sodium Current Kinetics through ROS and PKC Signalling in Induced Pluripotent Stem-Derived Cardiomyocytes from Brugada Syndrome Patient. Journal of Cardiovascular Development and Disease. 2022; 9(4):119. https://doi.org/10.3390/jcdd9040119
Chicago/Turabian StyleLiao, Zhenxing, Yingrui Li, Xuehui Fan, Zhen Yang, Ibrahim El-Battrawy, Xiaobo Zhou, and Ibrahim Akin. 2022. "Lipopolysaccharide Modifies Sodium Current Kinetics through ROS and PKC Signalling in Induced Pluripotent Stem-Derived Cardiomyocytes from Brugada Syndrome Patient" Journal of Cardiovascular Development and Disease 9, no. 4: 119. https://doi.org/10.3390/jcdd9040119
APA StyleLiao, Z., Li, Y., Fan, X., Yang, Z., El-Battrawy, I., Zhou, X., & Akin, I. (2022). Lipopolysaccharide Modifies Sodium Current Kinetics through ROS and PKC Signalling in Induced Pluripotent Stem-Derived Cardiomyocytes from Brugada Syndrome Patient. Journal of Cardiovascular Development and Disease, 9(4), 119. https://doi.org/10.3390/jcdd9040119