
Comparative Genomics Reveals Species-Specific Genes and Symbiotic Adaptations in Tricholoma matsutake
 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset Acquisition and Quality Assessment
2.2. Structural Annotation of Repetitive Elements, Genes, and RNAs
2.3. Synteny Analysis for T. matsutake Genomes
2.4. Functional Annotation of the Genes and Enrichment Tests
2.5. Phylogenomics, Orthology, and Gene Family Expansion/Contraction Analyses
2.6. Idenfication of a Trytophan Metabolism Pathway Gene by PCR Amplification
3. Results
3.1. Genome Quality and Metadata of the Available Agaricales Strains

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain | Isolated Location | No. of Scaffolds | Assembly Size (bp) | N50 (bp) | L50 | GC (%) | Repeat Size (bp) | Repeat Content (%) | Reference (GenBank Accession) |
|---|---|---|---|---|---|---|---|---|---|---|
| Tricholoma matsutake | NIFoS 2001 | Korea | 80 | 162,632,606 | 5,103,859 | 10 | 45.50 | 116,305,332 | 71.51 | Kang et al., 2023 [12] (GCA_026213095.1) |
| Tricholoma matsutake | KMCC04578 | Korea | 5255 | 189,030,256 | 93,406 | 589 | 45.25 | 99,821,686 | 69.22 | Min et al., 2020 [10] (GCA_002939025.2) |
| Tricholoma matsutake | MG52 | China | 58,276 | 155,036,874 | 9294 | 3971 | 45.35 | 108,421,248 | 71.23 | Li et al., 2018 [13] (GCA_003314635.1) |
| Tricholoma matsutake | NBRC 30605 | Japan | 88,884 | 131,718,925 | 2909 | 9526 | 45.46 | 88,187,244 | 67.19 | Unpublished (GCA_001652835.1) |
| Tricholoma matsutake | 945 | NA | 2545 | 175,758,422 | 320,875 | 167 | 45.40 | 104,910,411 | 69.01 | Miyauchi et al., 2020 [11] (GCA_014904895.1) |
| Tricholoma matsutake | Sample A | Japan | 13 | 161,040,721 | 12,376,747 | 6 | 45.50 | 115,538,238 | 71.74 | Kurokochi et al., 2023 [9] (GCA_026075535.2) |
| Tricholoma bakamatsutake | SF-Tf05 | Japan | 14 | 142,190,777 | 12,427,164 | 6 | 43.92 | 108,654,712 | 76.41 | Ichida et al., 2023 [38] (GCA_029959215.1) |
| Tricholoma bakamatsutake | MG51 | China | 51,308 | 140,676,125 | 9944 | 3260 | 43.93 | 106,664,015 | 77.08 | Li et al., 2018 [13] (GCA_003313665.1) |
| Tricholoma flavovirens | MG32 | China | 49,305 | 119,315,121 | 7637 | 3611 | 45.00 | 80,893,955 | 68.90 | Li et al., 2018 [13] (GCA_003313805.1) |
| Tricholoma sinoportentosum * | MG77 | China | 53,256 | 118,334,115 | 5062 | 6051 | 47.09 | 65,740,839 | 57.47 | Li et al., 2018 [13] (GCA_003314665.1) |
| Tricholoma saponaceum | MG146 | China | 14,500 | 55,708,857 | 11,929 | 1228 | 47.00 | 10,707,825 | 19.35 | Li et al., 2018 [13] (GCA_003313625.1) |
| Tricholoma albobrunneum ** | MG99 | China | 18,072 | 59,536,732 | 9454 | 1540 | 46.76 | 16,205,717 | 27.82 | Li et al., 2018 [13] (GCA_003521275.1) |
| Tricholoma hemisulphureum | KDTOL00252 | UK | 26 | 128,976,826 | 10,683,071 | 5 | 47.13 | 88,317,381 | 68.48 | Unpublished (GCA_964034925.1) |
| Tricholoma terreum | MG45 | China | 25,404 | 84,437,827 | 9877 | 2259 | 47.87 | 38,467,970 | 46.28 | Li et al., 2018 [13] (GCA_003316345.1) |
| Praearthromyces corneri *** | D33 | Malaysia | 17,094 | 49,805,319 | 10,870 | 876 | 47.44 | 11,299,287 | 22.73 | Unpublished (GCA_018854895.1) |
| Rugosomyces carneus | D47 | Netherlands | 39,733 | 58,963,700 | 3469 | 2576 | 47.02 | 15,608,583 | 26.69 | Unpublished (GCA_018221805.1) |
| Cystodermella granulosa | DBG-29008 | USA | 23,419 | 35,367,994 | 2089 | 4024 | 45.65 | 4,136,583 | 11.71 | Kraisitudomsook et al., 2024 [37] (GCA_036937035.1) |
| Flagelloscypha sp. | PMI 526 | NA | 397 | 72,728,845 | 1,132,480 | 18 | 46.84 | 9,103,480 | 12.52 | Unpublished (GCA_021399455.1) |
| Macrocystidia cucumis | KM177596 | UK | 204,508 | 46,746,859 | 15,511 | 599 | 48.76 | 4,791,427 | 10.33 | Unpublished (GCA_001179725.1) |
3.2. Evolutionary Relationship of the Agaricales Strains
3.3. Evolutionary Relationship of the Strains in Section Matsutake
3.4. Genome Statics of the Selected Agaricales Strains
3.5. Repeat Content within the Selected Strains
3.6. The Pan and Core Orthogroups of Selected Strains
3.7. The Gene Family Expansion and Contraction in Tricholoma Evolution
3.8. CAZyme, Secreted Protein, Effector, and Secondary Metabolite Gene Cluster Content in Selected Strains
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reschke, K.; Popa, F.; Yang, Z.L.; Kost, G. Diversity and taxonomy of Tricholoma species from Yunnan, China, and notes on species from Europe and North America. Mycologia 2018, 110, 1081–1109. [Google Scholar] [CrossRef] [PubMed]
- Benazza-Bouregba, M.; Savoie, J.-M.; Fortas, Z.; Billette, C. A new record of Tricholoma caligatum (Tricholomataceae) from North Africa with a discussion of related species. Phytotaxa 2016, 282, 119–128. [Google Scholar] [CrossRef]
- Cooper, J.A.; Park, D. The fungal genus Tricholomopsis (Agaricales) in New Zealand, including Tricholomopsis scabra sp. nov. Phytotaxa 2016, 288, 69–76. [Google Scholar] [CrossRef]
- Singer, R. Monographs of south American Basidiomycetes, especially those of the east slope of the Andes and Brazil. IX. Tricholoma in Brazil and Argentina. Darwiniana 1966, 14, 19–35. [Google Scholar]
- Sánchez-García, M.; Matheny, P.B. Is the switch to an ectomycorrhizal state an evolutionary key innovation in mushroom-forming fungi? A case study in the Tricholomatineae (Agaricales). Evolution 2017, 71, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.X.; Xu, X.; Cui, Y.Y.; Kost, G.; Wang, P.M.; Yang, Z.L. A fifty-locus phylogenetic analysis provides deep insights into the phylogeny of Tricholoma (Tricholomataceae, Agaricales). Persoonia 2023, 50, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, T.; Yamada, A.; Furukawa, H. Advances in the cultivation of the highly-prized ectomycorrhizal mushroom Tricholoma matsutake. Mycoscience 2020, 61, 49–57. [Google Scholar] [CrossRef]
- Aoki, W.; Bergius, N.; Kozlan, S.; Fukuzawa, F.; Okuda, H.; Murata, H.; Ishida, T.A.; Vaario, L.M.; Kobayashi, H.; Kalmiş, E.; et al. New findings on the fungal species Tricholoma matsutake from Ukraine, and revision of its taxonomy and biogeography based on multilocus phylogenetic analyses. Mycoscience 2022, 63, 197–214. [Google Scholar] [CrossRef]
- Kurokochi, H.; Tajima, N.; Sato, M.P.; Yoshitake, K.; Asakawa, S.; Isobe, S.; Shirasawa, K. Telomere-to-telomere genome assembly of matsutake (Tricholoma matsutake). DNA Res. 2023, 30, dsad006. [Google Scholar] [CrossRef]
- Min, B.; Yoon, H.; Park, J.; Oh, Y.L.; Kong, W.S.; Kim, J.G.; Choi, I.G. Unusual genome expansion and transcription suppression in ectomycorrhizal Tricholoma matsutake by insertions of transposable elements. PLoS ONE 2020, 15, e0227923. [Google Scholar] [CrossRef]
- Miyauchi, S.; Kiss, E.; Kuo, A.; Drula, E.; Kohler, A.; Sánchez-García, M.; Morin, E.; Andreopoulos, B.; Barry, K.W.; Bonito, G. Large-scale genome sequencing of mycorrhizal fungi provides insights into the early evolution of symbiotic traits. Nat. Commun. 2020, 11, 5125. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Bae, E.K.; Park, E.J.; Ka, K.H.; Son, M.R.; Kim, K.T.; Lee, J.W. Draft genome sequence for the symbiotic pine mushroom Tricholoma matsutake. Microbiol. Resour. Announc. 2023, 12, e0127122. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, S.; Ma, X.; Chen, W.; Zhang, J.; Duan, S.; Gao, Y.; Kui, L.; Huang, W.; Wu, P.; et al. The Genome Sequences of 90 Mushrooms. Sci. Rep. 2018, 8, 9982. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 25, 4.10.11–14.10.14. [Google Scholar] [CrossRef]
- Ou, S.; Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018, 176, 1410–1422. [Google Scholar] [CrossRef]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, i351–i358. [Google Scholar] [CrossRef]
- Bao, Z.; Eddy, S.R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef]
- Gabriel, L.; Brůna, T.; Hoff, K.J.; Ebel, M.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA. Genome Res. 2024, 34, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Brůna, T.; Lomsadze, A.; Borodovsky, M. GeneMark-EP+: Eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom. Bioinform. 2020, 2, lqaa026. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Barrnap 0.9: Rapid Ribosomal RNA Prediction. 2013. Available online: https://github.com/tseemann/barrnap (accessed on 31 May 2024).
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef] [PubMed]
- Alonge, M.; Lebeigle, L.; Kirsche, M.; Jenike, K.; Ou, S.; Aganezov, S.; Wang, X.; Lippman, Z.B.; Schatz, M.C.; Soyk, S. Automated assembly scaffolding using RagTag elevates a new tomato system for high-throughput genome editing. Genome Biol. 2022, 23, 258. [Google Scholar] [CrossRef]
- Marcais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [PubMed]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef]
- Zheng, J.; Ge, Q.; Yan, Y.; Zhang, X.; Huang, L.; Yin, Y. dbCAN3: Automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res. 2023, 51, W115–W121. [Google Scholar] [CrossRef]
- Sperschneider, J.; Dodds, P.N. EffectorP 3.0: Prediction of apoplastic and cytoplasmic effectors in fungi and oomycetes. Mol. Plant Microbe Interact. 2022, 35, 146–156. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J. antiSMASH 7.0: New and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 2023, 51, W46–W50. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Mendes, F.K.; Vanderpool, D.; Fulton, B.; Hahn, M.W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 2020, 36, 5516–5518. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; O’Meara, B.C. treePL: Divergence time estimation using penalized likelihood for large phylogenies. Bioinformatics 2012, 28, 2689–2690. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Suleski, M.; Craig, J.M.; Kasprowicz, A.E.; Sanderford, M.; Li, M.; Stecher, G.; Hedges, S.B. TimeTree 5: An expanded resource for species divergence times. Mol. Biol. Evol. 2022, 39, msac174. [Google Scholar] [CrossRef]
- Kraisitudomsook, N.; Ahrendt, S.; Riley, R.; LaButti, K.; Lipzen, A.; Daum, C.; Barry, K.; Grigoriev, I.V.; Rama, T.; Martin, F.; et al. On the origin of bird’s nest fungi: Phylogenomic analyses of fungi in the Nidulariaceae (Agaricales, Basidiomycota). Mol. Phylogenet. Evol. 2024, 193, 108010. [Google Scholar] [CrossRef]
- Ichida, H.; Murata, H.; Hatakeyama, S.; Yamada, A.; Ohta, A. Near-complete de novo assembly of Tricholoma bakamatsutake chromosomes revealed the structural divergence and differentiation of Tricholoma genomes. G3 Genes Genomes Genet. 2023, 13, jkad198. [Google Scholar] [CrossRef]
- van de Peppel, L.J.; Aime, M.C.; Læssøe, T.; Pedersen, O.S.; Coimbra, V.R.; Kuyper, T.W.; Stubbe, D.; Aanen, D.K.; Baroni, T.J. Four new genera and six new species of lyophylloid agarics (Agaricales, Basidiomycota) from three different continents. Mycol. Prog. 2022, 21, 85. [Google Scholar] [CrossRef]
- Frenkel, F.E.; Chaley, M.B.; Korotkov, E.V.; Skryabin, K.G. Evolution of tRNA-like sequences and genome variability. Gene 2004, 335, 57–71. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Hu, L.; Weng, J.; Wang, Z.; Huang, C.; Zhang, L. Effect and mechanism of Tricholoma matsutake extract combined with bakuchiol and ergothioneine on UVB-induced skin aging. J. Cosmet. Dermatol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Rajapitamahuni, S.; Kang, B.R.; Lee, T.K. Exploring the roles of arbuscular mycorrhizal fungi in plant–iron homeostasis. Agriculture 2023, 13, 1918. [Google Scholar] [CrossRef]
- Ying, X.; Ma, J.; Liang, Q.; Wang, Y.; Bai, G.; Luo, G. Identification and analysis of the constituents in an aqueous extract of Tricholoma matsutake by HPLC coupled with diode array detection/electrospray ionization mass spectrometry. J. Food Sci. 2013, 78, C1173–C1182. [Google Scholar] [CrossRef] [PubMed]
- Krauze-Baranowska, M.; Mardarowicz, M.; Wiwart, M.; Pobłocka, L.; Dynowska, M. Antifungal activity of the essential oils from some species of the genus Pinus. Z. Naturforsch. C 2002, 57, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Ringel, M.; Dimos, N.; Himpich, S.; Haack, M.; Huber, C.; Eisenreich, W.; Schenk, G.; Loll, B.; Brück, T. Biotechnological potential and initial characterization of two novel sesquiterpene synthases from Basidiomycota Coniophora puteana for heterologous production of δ-cadinol. Microb. Cell Fact. 2022, 21, 64. [Google Scholar] [CrossRef]
- Mohanta, T.K.; Bae, H. The diversity of fungal genome. Biol. Proced. Online 2015, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Vaario, L.M.; Yang, X.; Yamada, A. Biogeography of the Japanese gourmet fungus, Tricholoma matsutake: A review of the distribution and functional ecology of Matsutake. In Biogeography of Mycorrhizal Symbiosis; Springer: Cham, Switzerland, 2017; pp. 319–344. [Google Scholar]
- Lee, C.-Y.; Hong, O.-P.; Jung, M.-J.; Han, Y.-H. Effect of carbon sources and vitamins on mycelial growth of Tricholoma matsutake DGUM 26001. Kor. J. Mycol. 1997, 25, 226–232. [Google Scholar]
- Kusuda, M.; Ueda, M.; Konishi, Y.; Yamanaka, K.; Terashita, T.; Miyatake, K. Effects of carbohydrate substrate on the vegetative mycelial growth of an ectomycorrhizal mushroom, Tricholoma matsutake, isolated from Quercus. Mycoscience 2007, 48, 358–364. [Google Scholar] [CrossRef]
- Kusuda, M.; Nagai, M.; Hur, T.C.; Terashita, T.; Ueda, M. Purification and some properties of α-amylase from an ectomycorrhizal fungus, Tricholoma matsutake. Mycoscience 2003, 44, 311–317. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, X.; Li, G.; Wang, X. TORC1 signaling in Fungi: From yeasts to filamentous Fungi. Microorganisms 2023, 11, 218. [Google Scholar] [CrossRef]
- Chung, D.-Y.; Lee, K.-S.; Lee, J.-S.; Youn, Y.-N. Characteristics of a forest soil on pine mushroom habitat located in Ponghwa, Kyungbuk and Gansung, Kangwon. 1. Physical and chemical properties of O horizon and surface soil. Korean J. Soil Sci. Fert. 2008, 41, 206–213. [Google Scholar]
- Li, T.; Zhang, J.; Shen, T.; Shi, Y.D.; Yang, S.B.; Zhang, T.; Li, J.Q.; Wang, Y.Z.; Liu, H.G. Mineral element content in prized matsutake mushroom (Tricholoma matsutake) collected in China. Chem. Pap. 2013, 67, 672–676. [Google Scholar] [CrossRef]
- Li, Q.; Li, S.H.; Huang, W.L.; Liu, C.Y.; Xiong, C.; Li, X.L.; Zheng, L.Y. Mineral constituents of a prized edible mushroom (Tricholoma matsutake) and soils beneath the fruiting bodies from the production areas across China. J. Mt. Sci. 2016, 13, 2046–2052. [Google Scholar] [CrossRef]
- Looney, B.; Miyauchi, S.; Morin, E.; Drula, E.; Courty, P.E.; Kohler, A.; Kuo, A.; LaButti, K.; Pangilinan, J.; Lipzen, A. Evolutionary transition to the ectomycorrhizal habit in the genomes of a hyperdiverse lineage of mushroom-forming fungi. New Phytol. 2022, 233, 2294–2309. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.-M.; Ka, K.-H. Korean Tricholoma matsutake strains that promote mycorrhization and growth of pinus densiflora seedling. Kor. J. Mycol. 2001, 44, 155–165. [Google Scholar] [CrossRef]
- Madeira, F.; Madhusoodanan, N.; Lee, J.; Eusebi, A.; Niewielska, A.; Tivey, A.; Lopez, R.; Butcher, S. The EMBL-EBI Job Dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 2024, 52, W521–W525. [Google Scholar] [CrossRef]

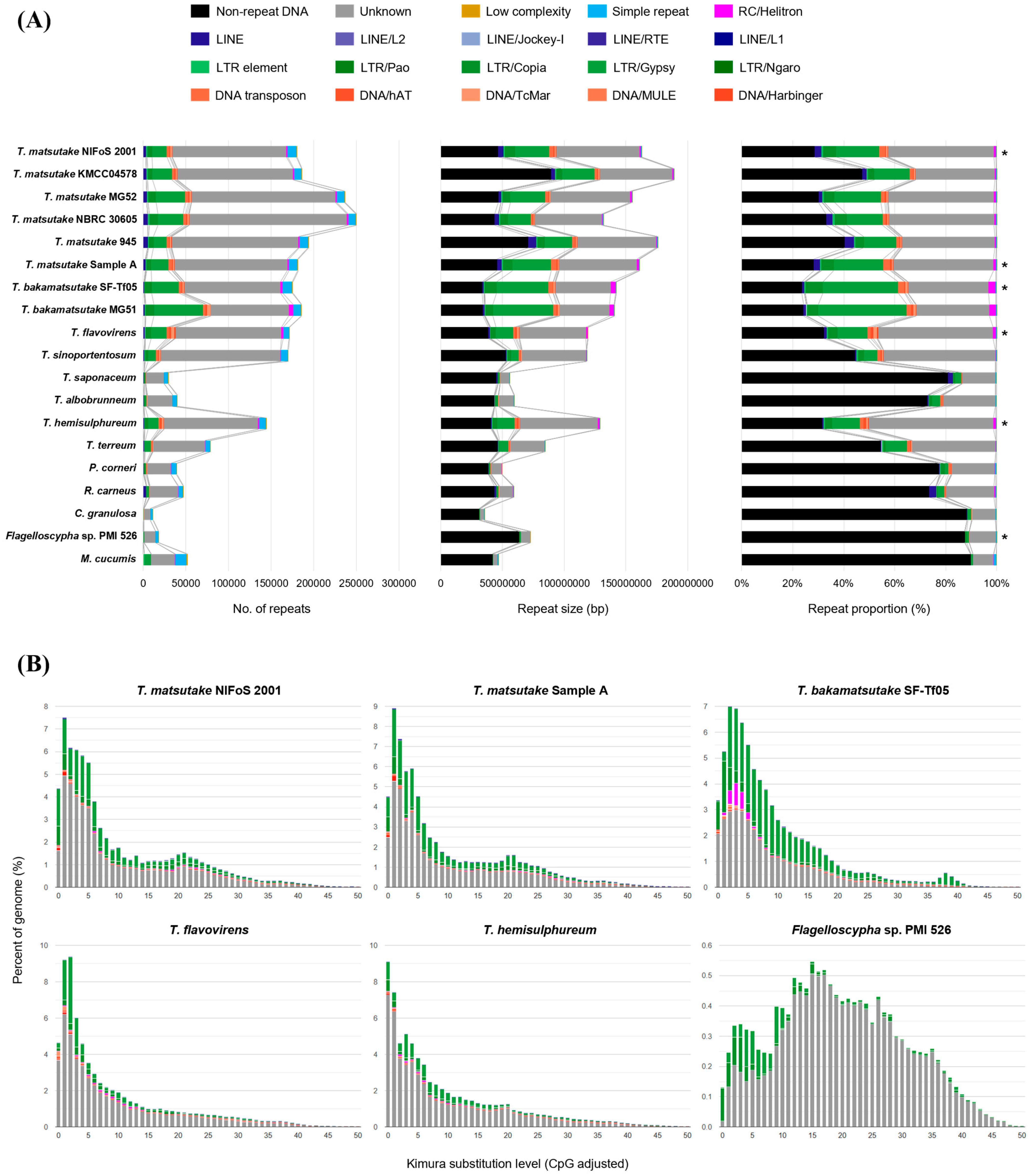
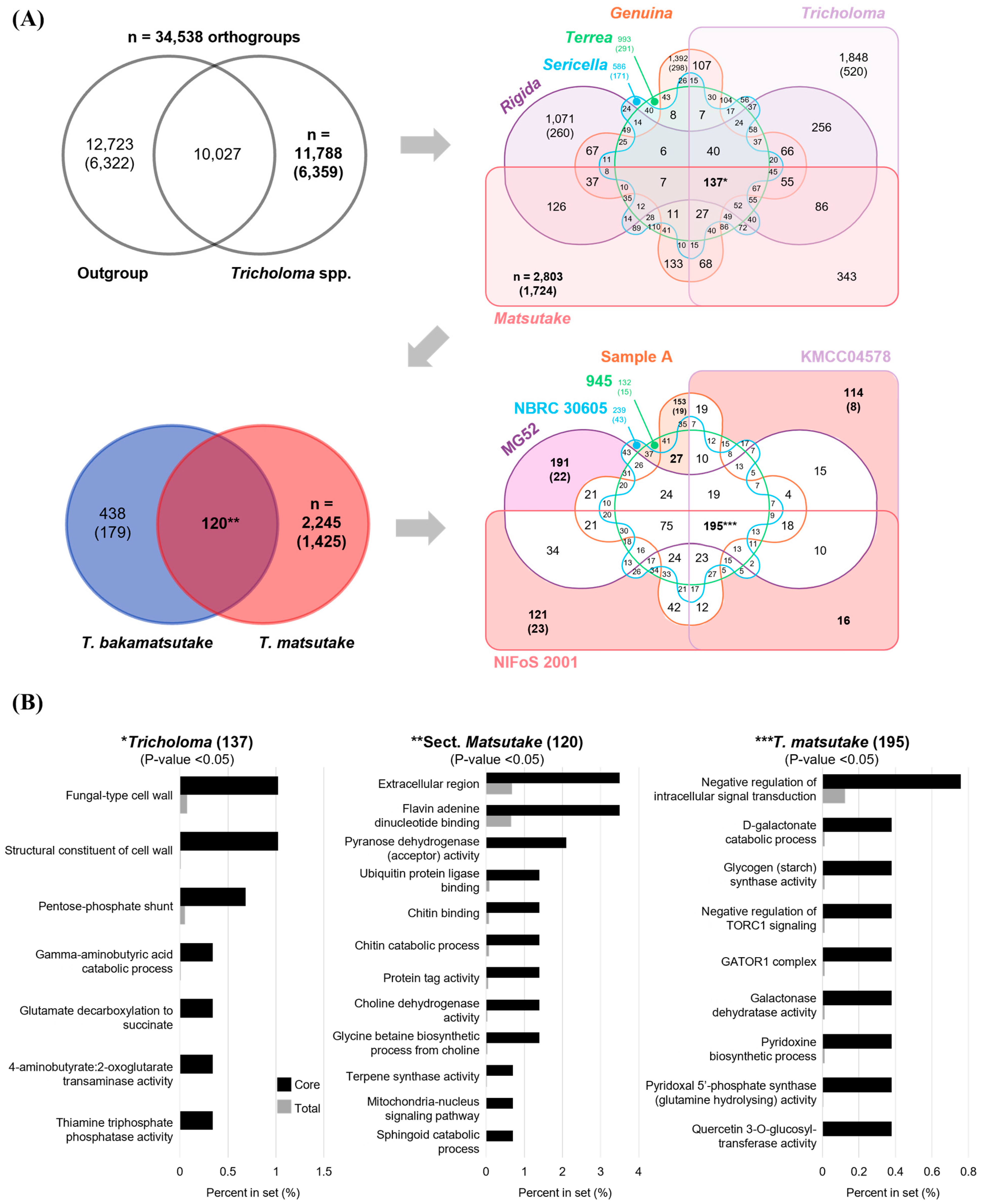


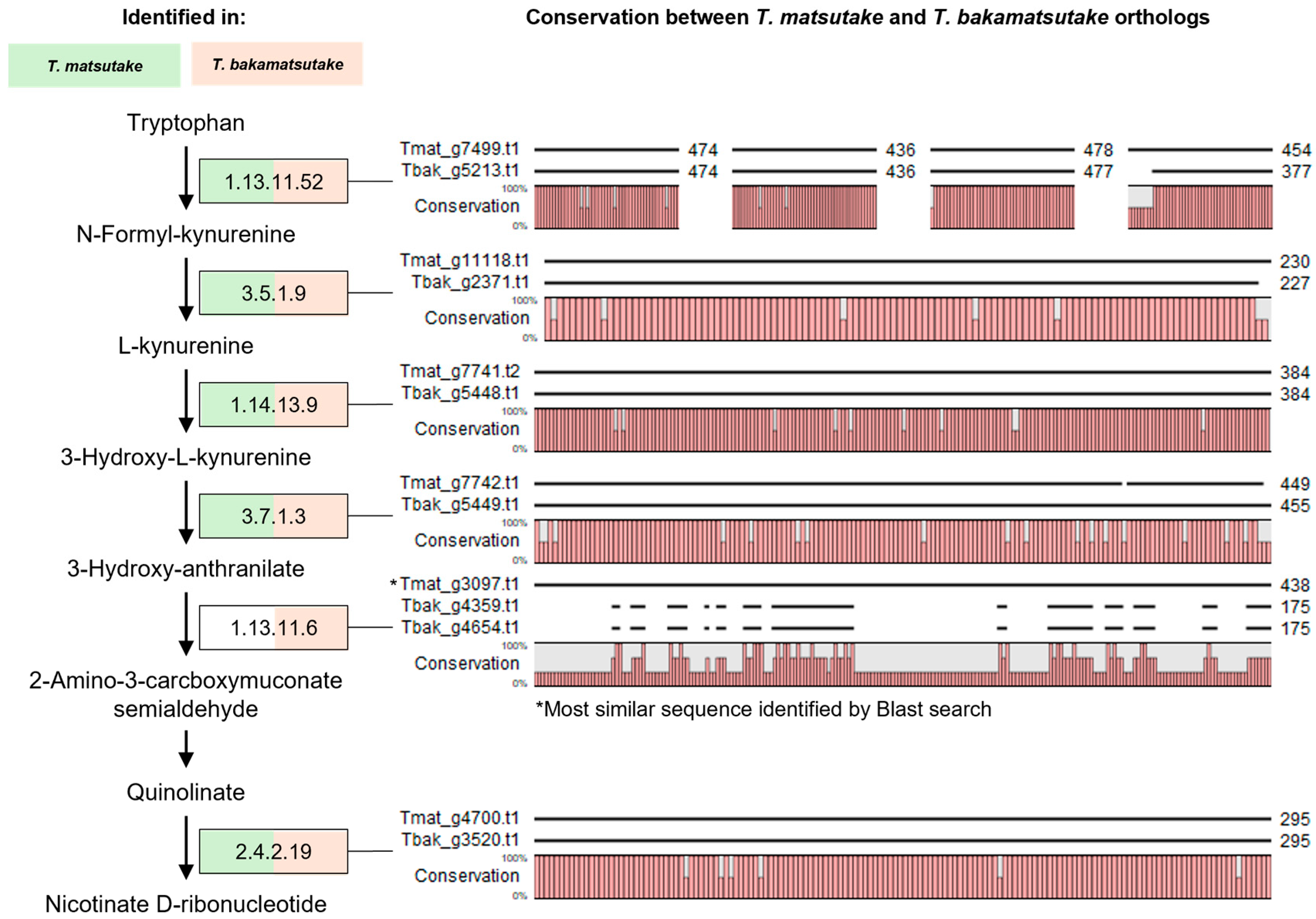
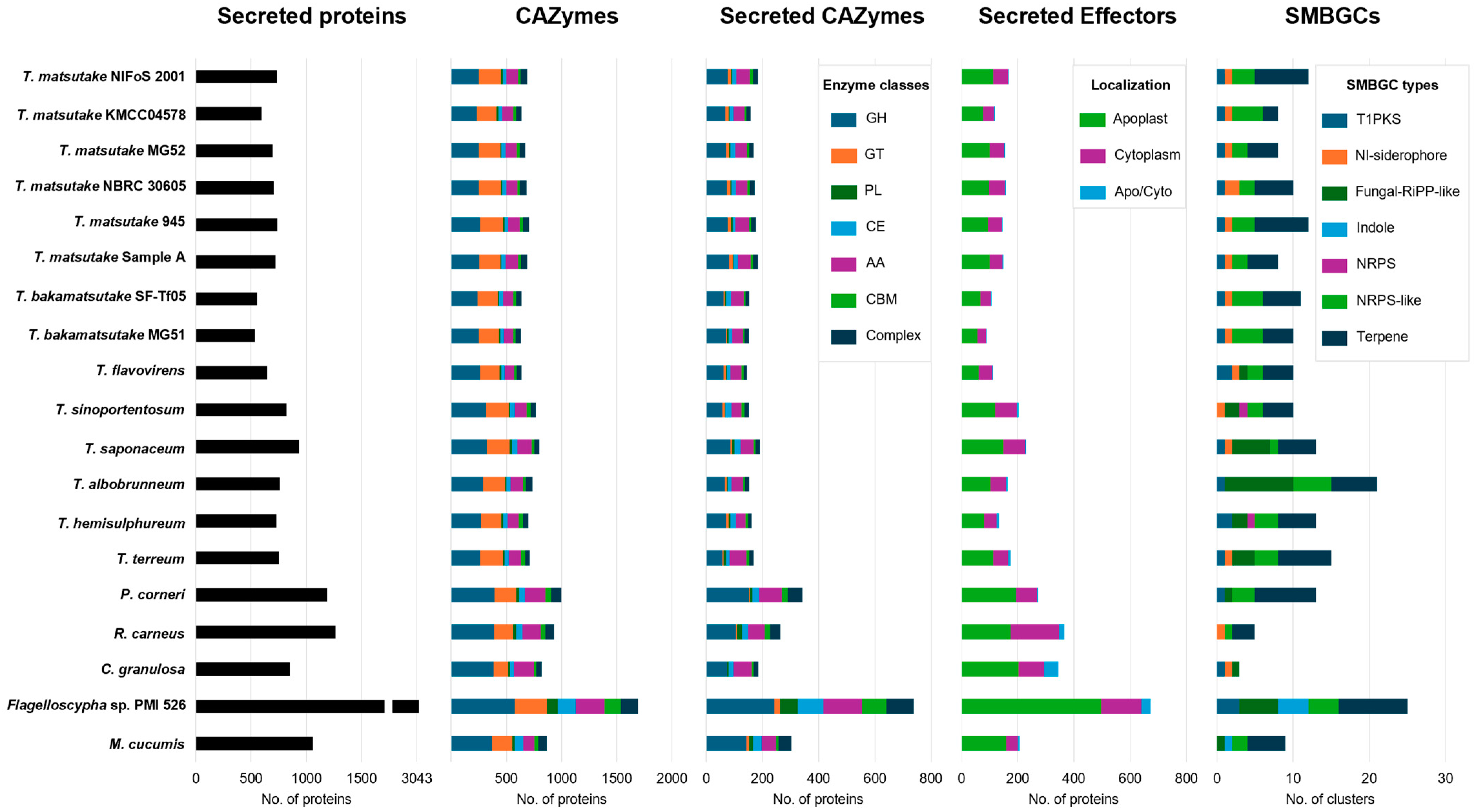
| Species | Strain | tRNAs | rRNAs | Protein-Coding Genes | Proteins | BLAST (%) | GO (%) | With EC no. (%) | InterPro (%) |
|---|---|---|---|---|---|---|---|---|---|
| Tricholoma matsutake | NIFoS 2001 | 277 | 25 | 14,657 | 17,257 | 16,410 (95.1) | 11,086 (64.2) | 3717 (21.5) | 10,890 (63.1) |
| Tricholoma matsutake | KMCC04578 | 275 | 4 | 11,449 | 13,364 | 12,862 (96.2) | 9318 (69.7) | 3444 (25.8) | 9230 (69.1) |
| Tricholoma matsutake | MG52 | 300 | 18 | 13,992 | 16,437 | 15,636 (95.1) | 10,739 (65.3) | 3774 (23.0) | 10,491 (63.8) |
| Tricholoma matsutake | NBRC 30605 | 304 | 9 | 13,931 | 16,356 | 15,559 (95.1) | 10,780 (65.9) | 3893 (23.8) | 10,325 (63.1) |
| Tricholoma matsutake | 945 | 281 | 3 | 14,442 | 17,048 | 16,292 (95.6) | 11,138 (65.3) | 3818 (22.4) | 11,028 (64.7) |
| Tricholoma matsutake | Sample A | 275 | 11 | 14,385 | 16,863 | 16,003 (94.9) | 10,820 (64.2) | 3639 (21.6) | 10,774 (63.9) |
| Tricholoma bakamatsutake | SF-Tf05 | 287 | 31 | 9697 | 11,071 | 10,646 (96.2) | 8464 (76.5) | 3145 (28.4) | 8486 (76.7) |
| Tricholoma bakamatsutake | MG51 | 283 | 45 | 9931 | 11,635 | 11,254 (96.7) | 8842 (76.0) | 3343 (28.7) | 8758 (75.3) |
| Tricholoma flavovirens | MG32 | 304 | 9 | 11,972 | 13,643 | 12,747 (93.4) | 9523 (69.8) | 3424 (25.1) | 9481 (69.5) |
| Tricholoma sinoportentosum | MG77 | 577 | 13 | 17,939 | 20,382 | 18,748 (92.0) | 13,799 (67.7) | 5037 (24.7) | 12,998 (63.8) |
| Tricholoma saponaceum | MG146 | 616 | 1 | 16,777 | 19,191 | 17,956 (93.6) | 13,533 (70.5) | 4827 (25.2) | 13,564 (70.7) |
| Tricholoma albobrunneum | MG99 | 516 | 22 | 15,597 | 17,587 | 16,261 (92.5) | 12,478 (71.0) | 4542 (25.8) | 12,271 (69.8) |
| Tricholoma hemisulphureum | KDTOL00252 | 305 | 1 | 12,460 | 14,103 | 13,017 (92.3) | 9819 (69.6) | 3523 (25.0) | 10,089 (71.5) |
| Tricholoma terreum | MG45 | 424 | 20 | 15,815 | 17,962 | 16,569 (92.2) | 12,503 (69.6) | 4286 (23.9) | 11,972 (66.7) |
| Praearthromyces corneri | D33 | 524 | 0 | 15,846 | 18,802 | 18,339 (97.5) | 12,342 (65.6) | 4662 (24.8) | 12,961 (78.9) |
| Rugosomyces carneus | D47 | 340 | 0 | 16,549 | 18,640 | 16,597 (89.0) | 13,393 (71.9) | 5428 (29.1) | 13,092 (70.2) |
| Cystodermella granulosa | DBG-29008 | 248 | 6 | 17,651 | 20,236 | 18,717 (92.5) | 15,841 (78.3) | 6974 (34.5) | 15,095 (74.6) |
| Flagelloscypha sp. | PMI 526 | 410 | 14 | 26,599 | 31,410 | 31,146 (99.2) | 18,012 (57.3) | 4205 (13.4) | 22,038 (70.2) |
| Macrocystidia cucumis | KM177596 | - * | 20 | 11,806 | 13,180 | 12,344 (93.7) | 10,079 (76.5) | 3441 (26.1) | 10,732 (81.4) |
| Total number of species/strains | 19 |
| Total number of proteins | 325,167 |
| Total number of orthogroups | 34,538 |
| Number of orthogroups with homologous proteins | 22,708 |
| Number of orthogroups with single protein (unassigned proteins) | 11,830 |
| Number of orthogroups with all species present | 4131 |
| Number of single-copy orthogroups | 204 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.H.; Bae, E.-K.; Hue, Y.; Choi, B.; Kang, M.-J.; Park, E.-J.; Kim, K.-T. Comparative Genomics Reveals Species-Specific Genes and Symbiotic Adaptations in Tricholoma matsutake. J. Fungi 2024, 10, 746. https://doi.org/10.3390/jof10110746
Kim JH, Bae E-K, Hue Y, Choi B, Kang M-J, Park E-J, Kim K-T. Comparative Genomics Reveals Species-Specific Genes and Symbiotic Adaptations in Tricholoma matsutake. Journal of Fungi. 2024; 10(11):746. https://doi.org/10.3390/jof10110746
Chicago/Turabian StyleKim, Jea Hyeoung, Eun-Kyung Bae, Yoeguang Hue, Byungheon Choi, Min-Jeong Kang, Eung-Jun Park, and Ki-Tae Kim. 2024. "Comparative Genomics Reveals Species-Specific Genes and Symbiotic Adaptations in Tricholoma matsutake" Journal of Fungi 10, no. 11: 746. https://doi.org/10.3390/jof10110746
APA StyleKim, J. H., Bae, E.-K., Hue, Y., Choi, B., Kang, M.-J., Park, E.-J., & Kim, K.-T. (2024). Comparative Genomics Reveals Species-Specific Genes and Symbiotic Adaptations in Tricholoma matsutake. Journal of Fungi, 10(11), 746. https://doi.org/10.3390/jof10110746