
Genome Sequencing and Metabolic Potential Analysis of Irpex lacteus


 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Collection of Strains and Culture Conditions
2.2. Genome Sequencing and Assembly
2.3. Gene Prediction and Annotation
2.4. CAZy and CYP Family in I. lacteus
2.5. Prediction of Gene Clusters Involved in Secondary Metabolites
2.6. Bioinformatics and Phylogenetic Analyses of STSs, PKSs, and P450s
2.7. MYB TFs Gene Family Analysis
3. Results
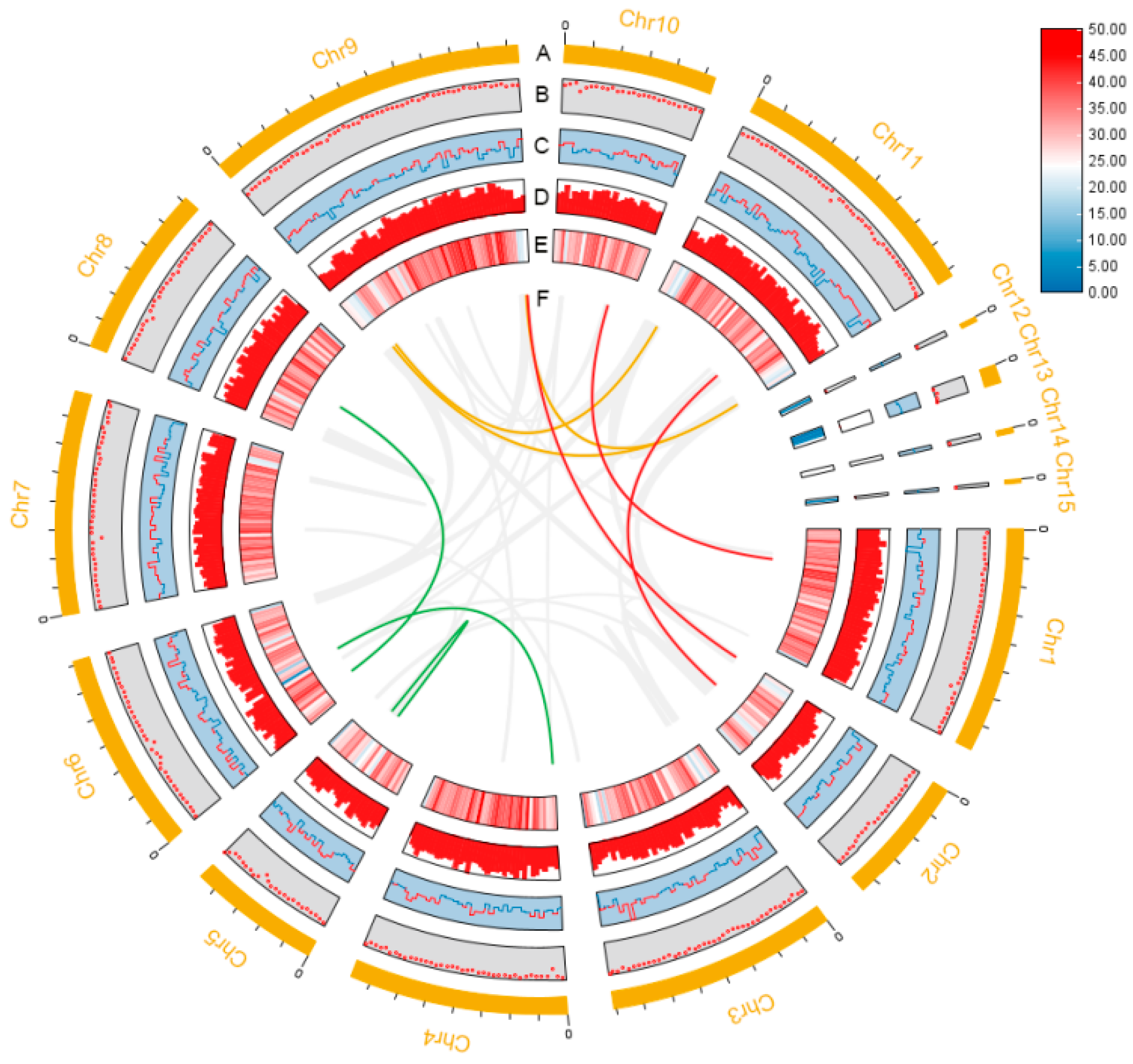
3.1. Genome Sequence Assembly and Annotation
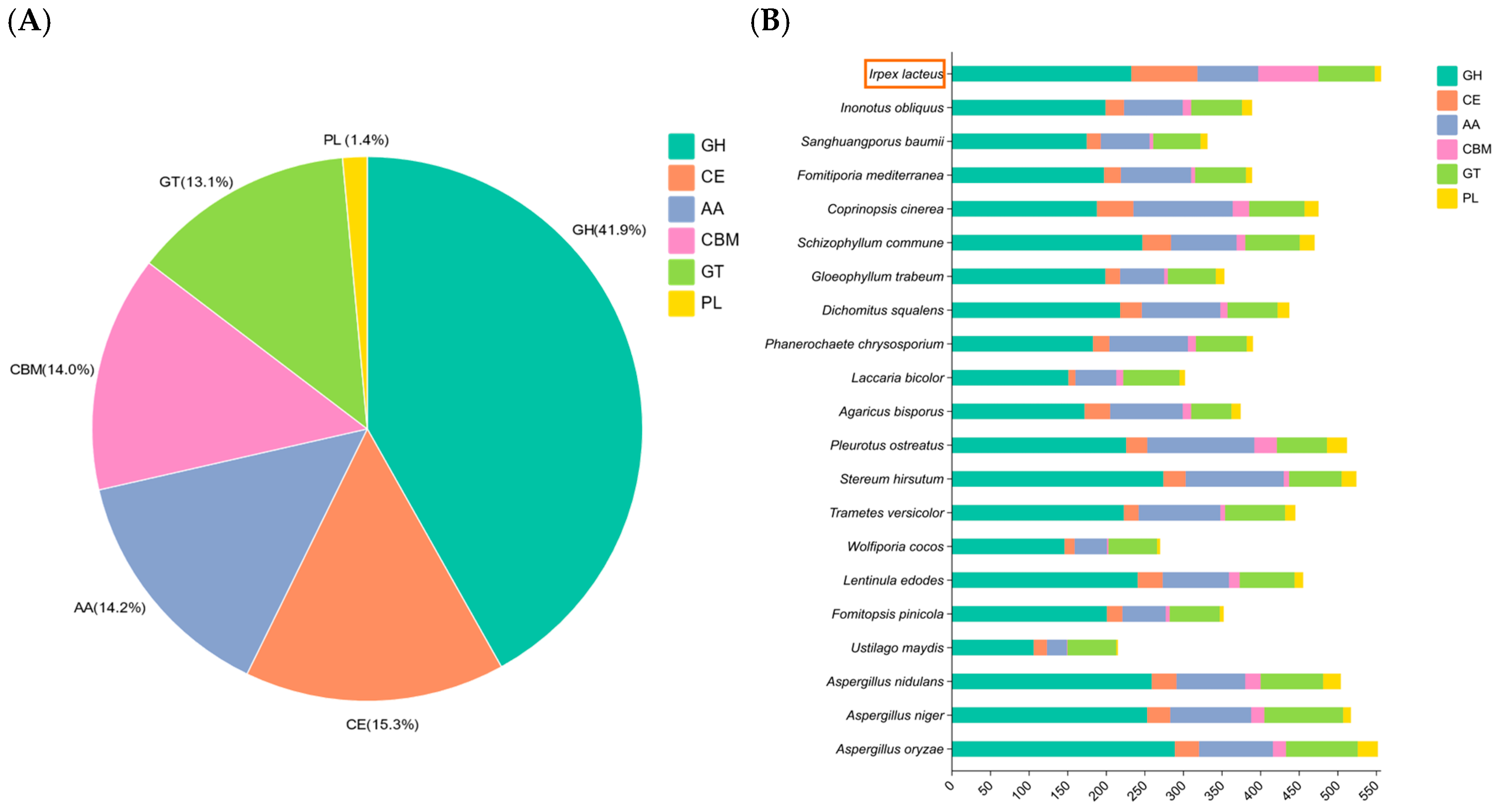
3.2. CAZymes Analysis
3.3. Mining of Biosynthetic Genes of Secondary Metabolic
3.3.1. Terpenoid Biosynthesis
3.3.2. Polyketide Biosynthesis
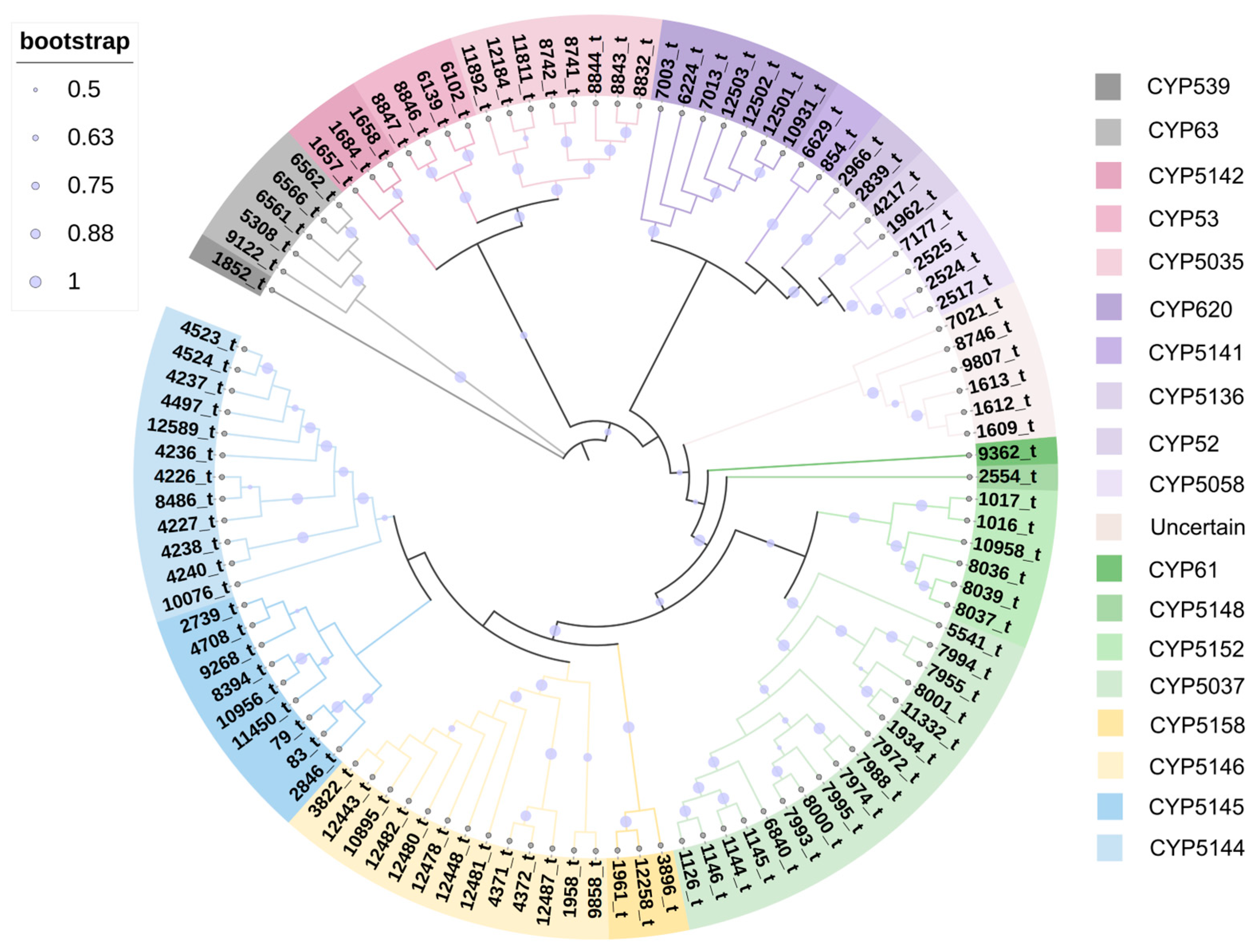
3.4. Cytochrome P450 Monooxygenase (CYP) Family Analysis
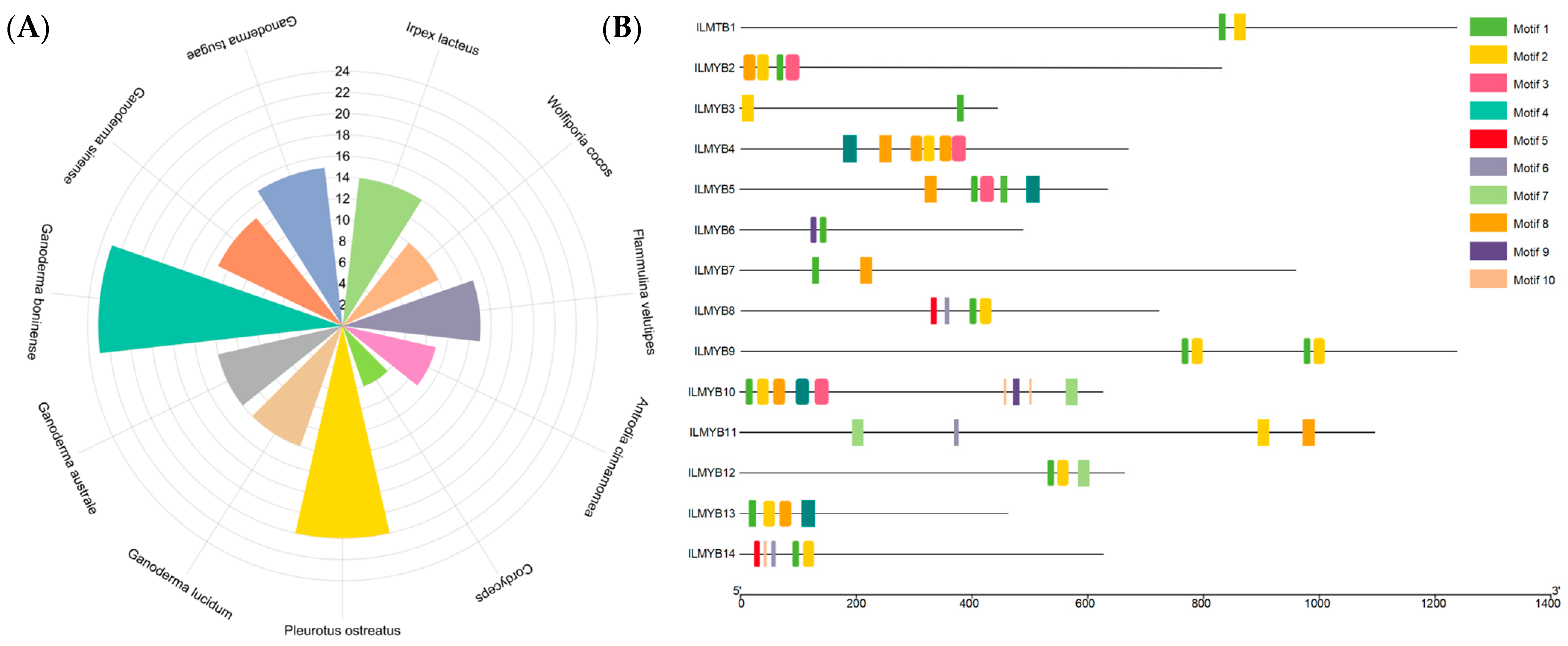
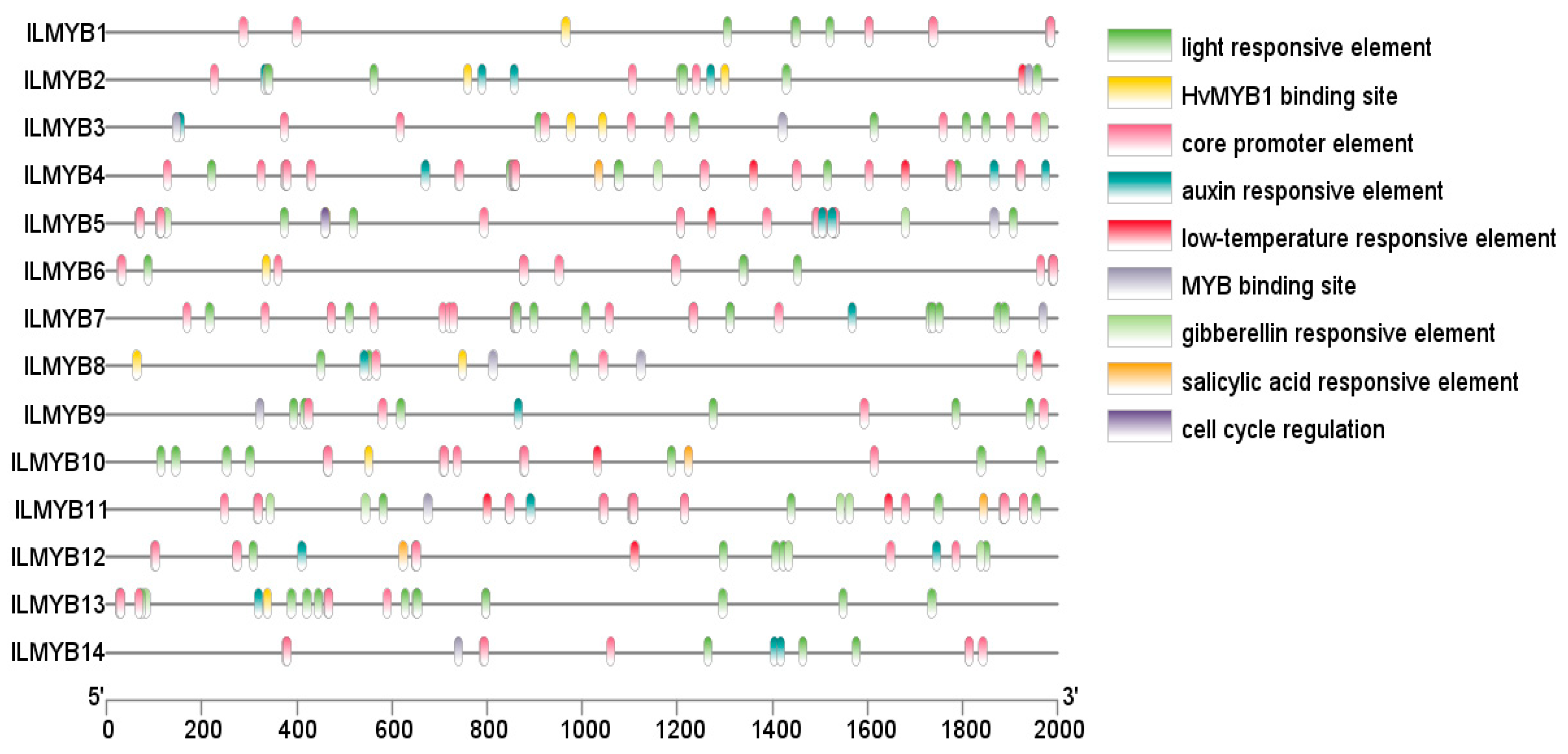
3.5. Genome-Wide Identification and Analysis of the MYB Gene Family
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dong, X.M.; Song, X.H.; Liu, K.B.; Dong, C.H. Prospect and current research status of medicinal fungus Irpex lacteus. Mycosystema 2017, 36, 28–34. [Google Scholar]
- Li, J.; Liu, Y.Y.; Hu, Z.Q.; Du, Z.W.; Wang, X.; Wang, X. Comparison of the effects of cultivation of Irpex lacteus in five different opening methods. Edible Med. Mushrooms 2023, 31, 401–405. [Google Scholar]
- Zhuang, L.; Guo, X.X.; Yu, S.T. Identification of ITS sequence and optimization of culture medium of a wild strain of Irpex lacteus. Edible Fungi 2022, 44, 27–29+32. [Google Scholar]
- Chen, R.; Feng, T.; Li, M.; Zhang, X.; He, J.; Hu, B.; Deng, Z.; Liu, T.; Liu, J.K.; Wang, X.; et al. Characterization of Tremulane Sesquiterpene Synthase from the Basidiomycete Irpex lacteus. Org. Lett. 2022, 24, 5669–5673. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, Z.H.; Isaka, M.; Liu, J.K.; Feng, T. Furan Derivatives and Polyketides from the Fungus Irpex lacteus. Nat. Prod. Bioprospect. 2021, 11, 215–222. [Google Scholar] [CrossRef]
- Wang, M.; Du, J.X.; Hui-Xiang, Y.; Dai, Q.; Liu, Y.P.; He, J.; Wang, Y.; Li, Z.H.; Feng, T.; Liu, J.K. Sesquiterpenoids from Cultures of the Basidiomycetes Irpex lacteus. J. Nat. Prod. 2020, 83, 1524–1531. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, Z.Z.; Yao, J.N.; Feng, T.; Li, Z.H.; Chen, H.P.; Liu, J.K. Irpeksins A-E, 1,10- seco-Eburicane-Type Triterpenoids from the Medicinal Fungus Irpex lacteus and Their Anti-NO Activity. J. Nat. Prod. 2018, 81, 2163–2168. [Google Scholar] [CrossRef]
- Duan, X.X.; Qin, D.; Song, H.C.; Gao, T.C.; Zuo, S.H.; Yan, X.; Wang, J.Q.; Ding, X.; Di, Y.T.; Dong, J.Y. Irpexlacte A-D, four new bioactive metabolites of endophytic fungus Irpex lacteus DR10-1 from the waterlogging tolerant plant Distylium chinense. Phytochem. Lett. 2019, 32, 151–156. [Google Scholar] [CrossRef]
- Dong, X.; Song, X.; Dong, C. Nutritional Requirements for Mycelial Growth of Milk-White Toothed Mushroom, Irpex lacteus (Agaricomycetes), in Submerged Culture. Int. J. Med. Mushrooms 2017, 19, 829–838. [Google Scholar] [CrossRef]
- Zhang, W.; Gao, G.P.; Cheng, R.C.; Qi, J.Y.; Wang, Y.; Wei, Z.H.; Zhao, L.W. Degradation Abilities of Irpex lacteus to Garden Tree Branches. J. Shenyang Agric. Univ. 2009, 40, 571–574. [Google Scholar]
- Malachova, K.; Rybkova, Z.; Sezimova, H.; Cerven, J.; Novotny, C. Biodegradation and detoxification potential of rotating biological contactor (RBC) with Irpex lacteus for remediation of dye-containing wastewater. Water Res. 2013, 47, 7143–7148. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Yu, H.; Song, L.; Zhang, J.; Weng, C.; Ma, F.; Zhang, X. The promoting effect of byproducts from Irpex lacteus on subsequent enzymatic hydrolysis of bio-pretreated cornstalks. Biotechnol. Biofuels 2011, 4, 37. [Google Scholar] [CrossRef]
- Cajthaml, T.; Erbanová, P.; Kollmann, A.; Novotný, C.; Sasek, V.; Mougin, C. Degradation of PAHs by ligninolytic enzymes of Irpex lacteus. Folia Microbiol. 2008, 53, 289–294. [Google Scholar] [CrossRef]
- Yang, J.Y.; Yang, Y.X.; Wang, X.Y.; Li, H.Q.; Zhang, B.H. Preliminary identification of an aroma-producing fungus strain and analysis of volatile compounds. Sci. Technol. Food Ind. 2015, 36, 197–200+205. [Google Scholar]
- Schadt, E.E.; Turner, S.; Kasarskis, A. A window into third-generation sequencing. Hum. Mol. Genet. 2010, 19, R227–R240. [Google Scholar] [CrossRef]
- Gong, W.; Wang, Y.; Xie, C.; Zhou, Y.; Zhu, Z.; Peng, Y. Whole genome sequence of an edible and medicinal mushroom, Hericium erinaceus (Basidiomycota, Fungi). Genomics 2020, 112, 2393–2399. [Google Scholar] [CrossRef]
- Duan, Y.; Han, H.; Qi, J.; Gao, J.M.; Xu, Z.; Wang, P.; Zhang, J.; Liu, C. Genome sequencing of Inonotus obliquus reveals insights into candidate genes involved in secondary metabolite biosynthesis. BMC Genom. 2022, 23, 314. [Google Scholar] [CrossRef]
- Zhao, C.; Feng, X.L.; Wang, Z.X.; Qi, J. The First Whole Genome Sequencing of Agaricus bitorquis and Its Metabolite Profiling. J Fungi 2023, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xu, J.; Liu, C.; Zhu, Y.; Nelson, D.R.; Zhou, S.; Li, C.; Wang, L.; Guo, X.; Sun, Y.; et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef]
- Dong, W.G.; Wang, Z.X.; Feng, X.L.; Zhang, R.Q.; Shen, D.Y.; Du, S.; Gao, J.M.; Qi, J. Chromosome-Level Genome Sequences, Comparative Genomic Analyses, and Secondary-Metabolite Biosynthesis Evaluation of the Medicinal Edible Mushroom Laetiporus sulphureus. Microbiol. Spectr. 2022, 10, e0243922. [Google Scholar] [CrossRef]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 2T5, 4–10. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated nr Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, C.L.M.; Chooi, Y.H. Synthaser: A CD-Search enabled Python toolkit for analysing domain architecture of fungal secondary metabolite megasynth(et)ases. Fungal Biol. Biotechnol. 2021, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Garron, M.L.; Henrissat, B. The continuing expansion of CAZymes and their families. Curr. Opin. Chem. Biol. 2019, 53, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Sista Kameshwar, A.K.; Qin, W. Comparative study of genome-wide plant biomass-degrading CAZymes in white rot, brown rot and soft rot fungi. Mycology 2018, 9, 93–105. [Google Scholar] [CrossRef]
- Rytioja, J.; Hildén, K.; Yuzon, J.; Hatakka, A.; de Vries, R.P.; Mäkelä, M.R. Plant-polysaccharide-degrading enzymes from Basidiomycetes. Microbiol. Mol. Biol. Rev. 2014, 78, 614–649. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Aguilar-Pontes, M.V.; Hainaut, M.; Henrissat, B.; Hildén, K.; Mäkelä, M.R.; de Vries, R.P. Comparative analysis of basidiomycete transcriptomes reveals a core set of expressed genes encoding plant biomass degrading enzymes. Fungal Genet. Biol. 2018, 112, 40–46. [Google Scholar] [CrossRef]
- Cragg, S.M.; Beckham, G.T.; Bruce, N.C.; Bugg, T.D.; Distel, D.L.; Dupree, P.; Etxabe, A.G.; Goodell, B.S.; Jellison, J.; McGeehan, J.E.; et al. Lignocellulose degradation mechanisms across the Tree of Life. Curr. Opin. Chem. Biol. 2015, 29, 108–119. [Google Scholar] [CrossRef]
- Wawrzyn, G.T.; Quin, M.B.; Choudhary, S.; López-Gallego, F.; Schmidt-Dannert, C. Draft genome of Omphalotus olearius provides a predictive framework for sesquiterpenoid natural product biosynthesis in Basidiomycota. Chem. Biol. 2012, 19, 772–783. [Google Scholar] [CrossRef]
- Flynn, C.M.; Schmidt-Dannert, C. Sesquiterpene Synthase-3-Hydroxy-3-Methylglutaryl Coenzyme A Synthase Fusion Protein Responsible for Hirsutene Biosynthesis in Stereum hirsutum. Appl. Environ. Microbiol. 2018, 84, e00036-18. [Google Scholar] [CrossRef]
- Wu, J.; Yang, X.; Duan, Y.; Wang, P.; Qi, J.; Gao, J.M.; Liu, C. Biosynthesis of Sesquiterpenes in Basidiomycetes: A Review. J. Fungi 2022, 8, 913. [Google Scholar] [CrossRef]
- Lackner, G.; Misiek, M.; Braesel, J.; Hoffmeister, D. Genome mining reveals the evolutionary origin and biosynthetic potential of basidiomycete polyketide synthases. Fungal Genet. Biol. 2012, 49, 996–1003. [Google Scholar] [CrossRef]
- Hoffmeister, D.; Keller, N.P. Natural products of filamentous fungi: Enzymes, genes, and their regulation. Nat. Prod. Rep. 2007, 24, 393–416. [Google Scholar] [CrossRef] [PubMed]
- Ishiuchi, K.; Nakazawa, T.; Ookuma, T.; Sugimoto, S.; Sato, M.; Tsunematsu, Y.; Ishikawa, N.; Noguchi, H.; Hotta, K.; Moriya, H.; et al. Establishing a new methodology for genome mining and biosynthesis of polyketides and peptides through yeast molecular genetics. ChemBioChem 2012, 13, 846–854. [Google Scholar] [CrossRef]
- Lackner, G.; Bohnert, M.; Wick, J.; Hoffmeister, D. Assembly of melleolide antibiotics involves a polyketide synthase with cross-coupling activity. Chem. Biol. 2013, 20, 1101–1106. [Google Scholar] [CrossRef]
- Yu, P.W.; Chang, Y.C.; Liou, R.F.; Lee, T.H.; Tzean, S.S. pks63787, a Polyketide Synthase Gene Responsible for the Biosynthesis of Benzenoids in the Medicinal Mushroom Antrodia cinnamomea. J. Nat. Prod. 2016, 79, 1485–1491. [Google Scholar] [CrossRef]
- Han, H.; Yu, C.; Qi, J.; Wang, P.; Zhao, P.; Gong, W.; Xie, C.; Xia, X.; Liu, C. High-efficient production of mushroom polyketide compounds in a platform host Aspergillus oryzae. Microb. Cell Fact. 2023, 22, 60. [Google Scholar] [CrossRef]
- Reyes-Fernández, E.Z.; Shi, Y.M.; Grün, P.; Bode, H.B.; Bölker, M. An Unconventional Melanin Biosynthesis Pathway in Ustilago maydis. Appl. Environ. Microbiol. 2021, 87, e01510-20. [Google Scholar] [CrossRef]
- Kuang, Y.; Li, B.; Wang, Z.; Qiao, X.; Ye, M. Terpenoids from the medicinal mushroom Antrodia camphorata: Chemistry and medicinal potential. Nat. Prod. Rep. 2021, 38, 83–102. [Google Scholar] [CrossRef]
- Araki, Y.; Awakawa, T.; Matsuzaki, M.; Cho, R.; Matsuda, Y.; Hoshino, S.; Shinohara, Y.; Yamamoto, M.; Kido, Y.; Inaoka, D.K.; et al. Complete biosynthetic pathways of ascofuranone and ascochlorin in Acremonium egyptiacum. Proc. Natl. Acad. Sci. USA 2019, 116, 8269–8274. [Google Scholar] [CrossRef]
- Shin, J.; Kim, J.E.; Lee, Y.W.; Son, H. Fungal Cytochrome P450s and the P450 Complement (CYPome) of Fusarium graminearum. Toxins 2018, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Crešnar, B.; Petrič, S. Cytochrome P450 enzymes in the fungal kingdom. Biochim. Biophys. Acta 2011, 1814, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Guo, J.; Cheng, F.; Li, S. Cytochrome P450 enzymes in fungal natural product biosynthesis. Nat. Prod. Rep. 2021, 38, 1072–1099. [Google Scholar] [CrossRef]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Wang, B.; Luo, Q.; Li, Y.; Yin, L.; Zhou, N.; Li, X.; Gan, J.; Dong, A. Structural insights into target DNA recognition by R2R3-MYB transcription factors. Nucleic Acids Res. 2020, 48, 460–471. [Google Scholar] [CrossRef]
- Du, H.; Liang, Z.; Zhao, S.; Nan, M.G.; Tran, L.S.; Lu, K.; Huang, Y.B.; Li, J.N. The Evolutionary History of R2R3-MYB Proteins Across 50 Eukaryotes: New Insights Into Subfamily Classification and Expansion. Sci. Rep. 2015, 5, 11037. [Google Scholar] [CrossRef]
- Zhou, Y.; Ness, S.A. Myb proteins: Angels and demons in normal and transformed cells. Front. Biosci. Landmark 2011, 16, 1109–1131. [Google Scholar] [CrossRef]
- Bessa, M.; Joaquin, M.; Tavner, F.; Saville, M.K.; Watson, R.J. Regulation of the cell cycle by B-Myb. Blood Cells Mol. Dis. 2001, 27, 416–421. [Google Scholar] [CrossRef]
- Tamagnone, L.; Merida, A.; Parr, A.; Mackay, S.; Culianez-Macia, F.A.; Roberts, K.; Martin, C. The AmMYB308 and AmMYB330 transcription factors from antirrhinum regulate phenylpropanoid and lignin biosynthesis in transgenic tobacco. Plant Cell 1998, 10, 135–154. [Google Scholar] [CrossRef]
- Arratia-Quijada, J.; Sánchez, O.; Scazzocchio, C.; Aguirre, J. FlbD, a Myb transcription factor of Aspergillus nidulans, is uniquely involved in both asexual and sexual differentiation. Eukaryot. Cell 2012, 11, 1132–1142. [Google Scholar] [CrossRef]
- Sarikaya Bayram, Ö.; Latgé, J.P.; Bayram, Ö. MybA, a new player driving survival of the conidium of the human pathogen Aspergillus fumigatus. Curr. Genet. 2018, 64, 141–146. [Google Scholar] [CrossRef]
- Li, X.; Li, W.J.; Wang, F.; Tang, L.; Qian, Z.M.; Dong, C.H. Identification of the MYB family of transcription factors and gene expression analysis during the growth and development of Chinese cordyceps. Mycosystema 2019, 38, 2174–2182. [Google Scholar]
- Wang, L.; Huang, Q.; Zhang, L.; Wang, Q.; Liang, L.; Liao, B. Genome-Wide Characterization and Comparative Analysis of MYB Transcription Factors in Ganoderma Species. G3 Genes Genomes Genet. 2020, 10, 2653–2660. [Google Scholar] [CrossRef]
- Deng, B.; Liu, Z.Q.; Yuan, X.W.; Liu, J.Y.; Meng, J.L.; Chang, M.C.; Feng, C.P. Identification of MYB Transcription Factor Family Members of Flammulina filiformis and Analysis of Their Expression Pattern During Fruiting Body Development. Acta Edulis Fungi 2021, 37, 1–11. [Google Scholar]
- Zhang, Z.; Wang, Y.; Luo, M.; Wang, J.; Yang, Y.M.; Zheng, Y. Genome-wide Identification and Analysis of MYB Transcription Factors of Antrodia cinnamomea. Mol. Plant Breed. 2022, 20, 4634–4641. [Google Scholar]
- Chen, H.Y.; Dong, S.Y.; Guo, M.X.; Luo, H.M. Genome-wide characterization and expression analysis of MYB transcriptionfactor gene family in Poria cocos. Chin. Tradit. Herb. Drugs 2023, 54, 245–253. [Google Scholar]
- Yuan, H.; Liu, Z.; Guo, L.; Hou, L.; Meng, J.; Chang, M. Function of Transcription Factors PoMYB12, PoMYB15, and PoMYB20 in Heat Stress and Growth of Pleurotus ostreatus. Int. J. Mol. Sci. 2023, 24, 13559. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, Z.Z.; Feng, T.; Li, Z.H.; Chen, H.P.; Liu, J.K. Triterpenes with unusual modifications from the fruiting bodies of the medicinal fungus Irpex lacteus. Int. J. Mol. Sci. 2023, 24, 21–28. [Google Scholar] [CrossRef]
- Ding, J.H.; Li, Z.H.; Feng, T.; Liu, J.K. Tremulane sesquiterpenes from cultures of the basidiomycete Irpex lacteus. Fitoterapia 2018, 125, 245–248. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, Z.Z.; Li, Z.H.; Feng, T.; Chen, H.P.; Liu, J.K. Irpexoates A-D, Four Triterpenoids with Malonyl Modifications from the Fruiting Bodies of the Medicinal Fungus Irpex lacteus. Nat. Prod. Bioprospect 2018, 8, 171–176. [Google Scholar] [CrossRef]
- Luo, H.Z.; Jiang, H.; Sun, B.; Wang, Z.N.; Jia, A.Q. Sesquiterpenoids and furan derivatives from the Orychophragmus violaceus (L.) O.E. Schulz endophytic fungus Irpex lacteus OV38. Phytochemistry 2022, 194, 112996. [Google Scholar] [CrossRef]
- Yao, M.; Li, W.; Duan, Z.; Zhang, Y.; Jia, R. Genome sequence of the white-rot fungus Irpex lacteus F17, a type strain of lignin degrader fungus. Stand. Genom. Sci. 2017, 12, 55. [Google Scholar] [CrossRef]
- Qin, X.; Su, X.; Luo, H.; Ma, R.; Yao, B.; Ma, F. Deciphering lignocellulose deconstruction by the white rot fungus Irpex lacteus based on genomic and transcriptomic analyses. Biotechnol. Biofuels 2018, 11, 58. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.F.; Xiao, H.; Zhong, J.J. Biosynthesis of a novel ganoderic acid by expressing CYP genes from Ganoderma lucidum in Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2022, 106, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Jiang, C.; Wang, Q.; Fang, Y.; Wang, J.; Wang, M.; Xiao, H. Biosynthesis of mushroom-derived type II ganoderic acids by engineered yeast. Nat. Commun. 2022, 13, 7740. [Google Scholar] [CrossRef]
- Fessner, N.D.; Nelson, D.R.; Glieder, A. Evolution and enrichment of CYP5035 in Polyporales: Functionality of an understudied P450 family. Appl. Microbiol. Biotechnol. 2021, 105, 6779–6792. [Google Scholar] [CrossRef]
- Lee, S.; Völz, R.; Song, H.; Harris, W.; Lee, Y.H. Characterization of the MYB Genes Reveals Insights into Their Evolutionary Conservation, Structural Diversity, and Functional Roles in Magnaporthe oryzae. Front. Microbiol. 2021, 12, 721530. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Value |
|---|---|
| Total length (bp) | 41,831,088 |
| Contigs | 55 |
| Scaffolds | 55 |
| N50 (bp) | 3,951,072 |
| N90 (bp) | 2,327,118 |
| L50 | 5 |
| L90 | 10 |
| GC% | 49.82 |
| Characteristics | Value |
|---|---|
| CDS number | 13,135 |
| CDS total length | 20,105,286 bp |
| CDS density | 0.314 genes per kb |
| CDS average length | 1530 bp |
| Intergenetic region length | 21,725,802 bp |
| CDS/Genome (coding percentage) | 48.10% |
| Intergenetic length/Genome | 51.90% |
| GC content in gene region | 53.70% |
| Cluster No. | Location | Start (bp) | End (bp) | Core Gene ID | Core Gene Type |
|---|---|---|---|---|---|
| 1 | Chr2 | 1,966,880 | 1,968,009 | 1945_t | Terpene |
| 2 | Chr3 | 1,287,295 | 1,297,241 | 2437_t | PKS |
| 3 | Chr4 | 1,179,836 | 1,184,996 | 3714_t | NRPS-like |
| 4 | Chr5 | 1,262,257 | 1,266,817 | 5076_t | NRPS-like |
| 5 | Chr6 | 1,742,732 | 1,745,523 | 5909_t | Terpene |
| 6 | Chr6 | 2,091,664 | 2,095,641 | 6013_t | NRPS-like |
| 7 | Chr6 | 2,530,444 | 2,532,296 | 6158_t | Terpene |
| 8 | Chr6 | 2,907,033 | 2,908,352 | 6273_t | Terpene |
| 9 | Chr7 | 1,027,290 | 1,028,582 | 6816_t | Terpene |
| 10 | Chr7 | 1,562,469 | 1,566,947 | 7012_t | NRPS-like |
| 11 | Chr7 | 2,469,431 | 2,470,534 | 7340_t | Terpene |
| 12 | Chr7 | 2,471,371 | 2,472,669 | 7341_t | Terpene |
| 13 | Chr7 | 3,746,009 | 3,750,647 | 7760_t | PKS |
| 14 | Chr7 | 3,765,445 | 3,766,804 | 7766_t | PKS |
| 15 | Chr8 | 597,512 | 601,542 | 8028_t | NRPS-like |
| 16 | Chr8 | 705,376 | 709,477 | 8060_t | NRPS-like |
| 17 | Chr8 | 1,189,829 | 1,192,243 | 8219_t | Terpene |
| 18 | Chr8 | 2,059,227 | 2,060,877 | 8523_t | Terpene |
| 19 | Chr9 | 763,448 | 767,556 | 9069_t | NRPS-like |
| 20 | Chr10 | 637,671 | 639,055 | 10957_t | Terpene |
| 21 | Chr10 | 1,622,548 | 1,629,357 | 11290_t | PKS |
| 22 | Chr10 | 2,367,015 | 2,368,086 | 11546_t | Terpene |
| 23 | Chr10 | 2,369,863 | 2,371,065 | 11547_t | Terpene |
| 24 | Chr10 | 2,422,443 | 2,423,554 | 11558_t | Terpene |
| 25 | Chr10 | 2,425,282 | 2,427,793 | 11559_t | Terpene |
| 26 | Chr11 | 2,261,230 | 2,265,279 | 12363_t | NRPS-like |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Duan, Y.; Zhang, M.; Liang, C.; Li, W.; Liu, C.; Ye, Y. Genome Sequencing and Metabolic Potential Analysis of Irpex lacteus. J. Fungi 2024, 10, 846. https://doi.org/10.3390/jof10120846
Wang Y, Duan Y, Zhang M, Liang C, Li W, Liu C, Ye Y. Genome Sequencing and Metabolic Potential Analysis of Irpex lacteus. Journal of Fungi. 2024; 10(12):846. https://doi.org/10.3390/jof10120846
Chicago/Turabian StyleWang, Yue, Yingce Duan, Menghan Zhang, Chaoqin Liang, Wenli Li, Chengwei Liu, and Ying Ye. 2024. "Genome Sequencing and Metabolic Potential Analysis of Irpex lacteus" Journal of Fungi 10, no. 12: 846. https://doi.org/10.3390/jof10120846
APA StyleWang, Y., Duan, Y., Zhang, M., Liang, C., Li, W., Liu, C., & Ye, Y. (2024). Genome Sequencing and Metabolic Potential Analysis of Irpex lacteus. Journal of Fungi, 10(12), 846. https://doi.org/10.3390/jof10120846