
Protective Effect of Indole-3-Aldehyde in Murine COVID-19-Associated Pulmonary Aspergillosis
 , , , , , ,
, , , , , ,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice, Infections, and Treatments
2.2. SiRNA Design and Delivery
2.3. TUNEL Staining
2.4. Real-Time PCR
2.5. Cytokine Determination by ELISA
2.6. Cells, Infection, and Treatments
2.7. Plaque Reduction Assay
2.8. Cytopathic Effect Inhibition Assay
2.9. Statistical Analysis
3. Results
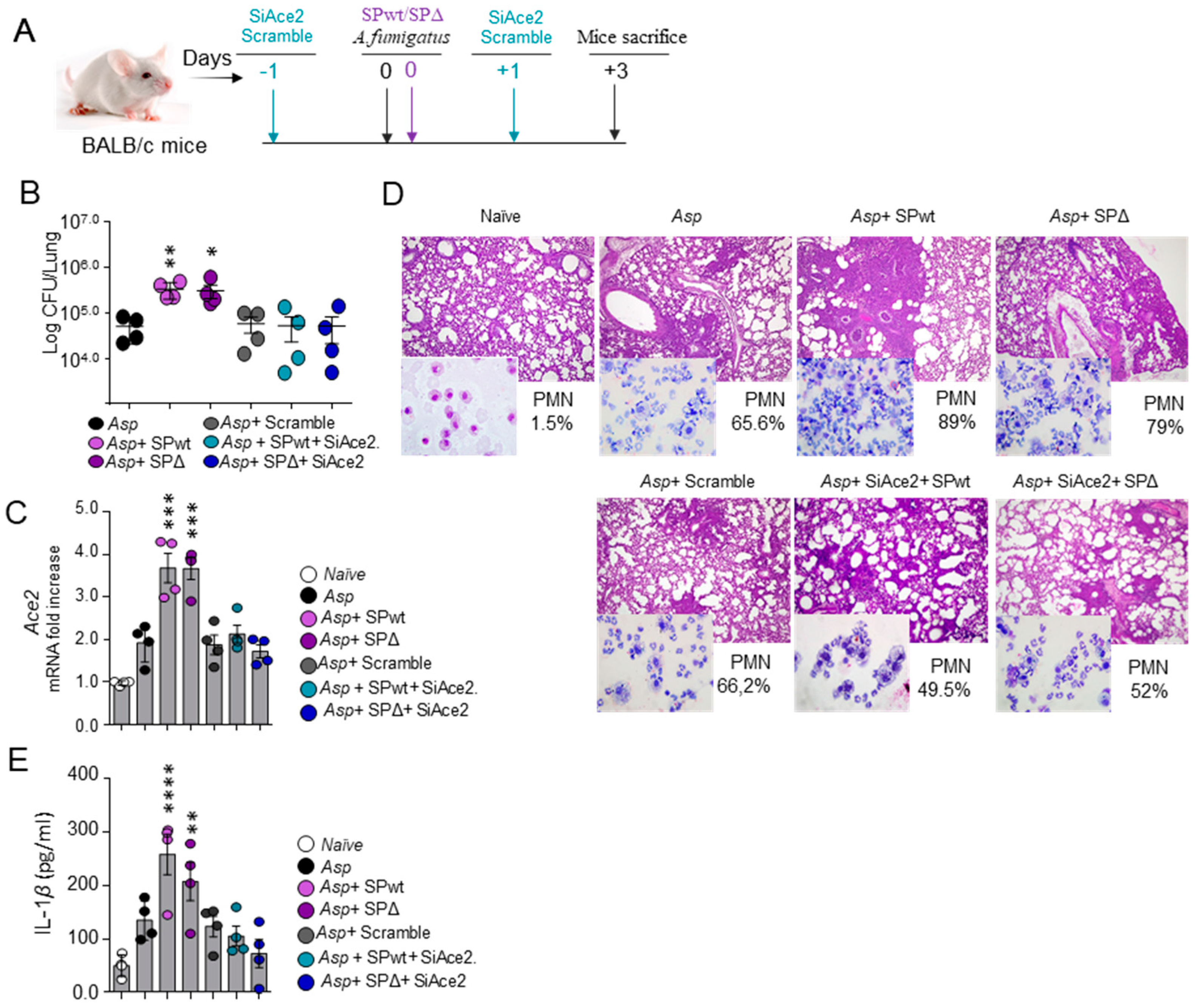
3.1. SARS-CoV-2 Spike Protein Worsens Aspergillus Infection in a Murine Model of CAPA
3.2. 3-IAld Protects against CAPA
3.3. 3-IAld Restores Mucosal Homeostasis in CAPA via the Aryl Hydrocarbon Receptor
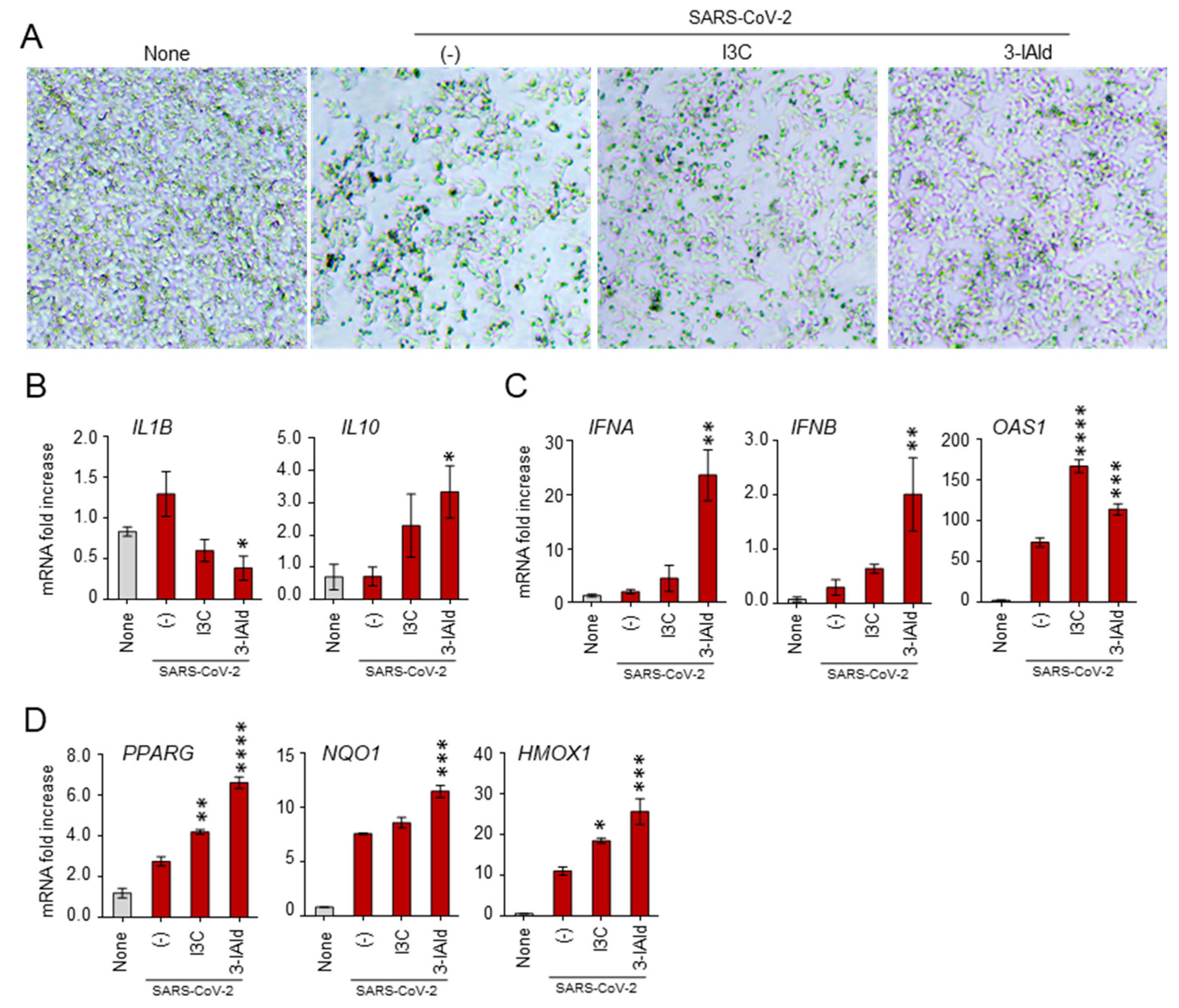
3.4. 3-IAld Counteracts the SARS-CoV-2 Immunomodulatory Effects in Nasal Epithelial Cells
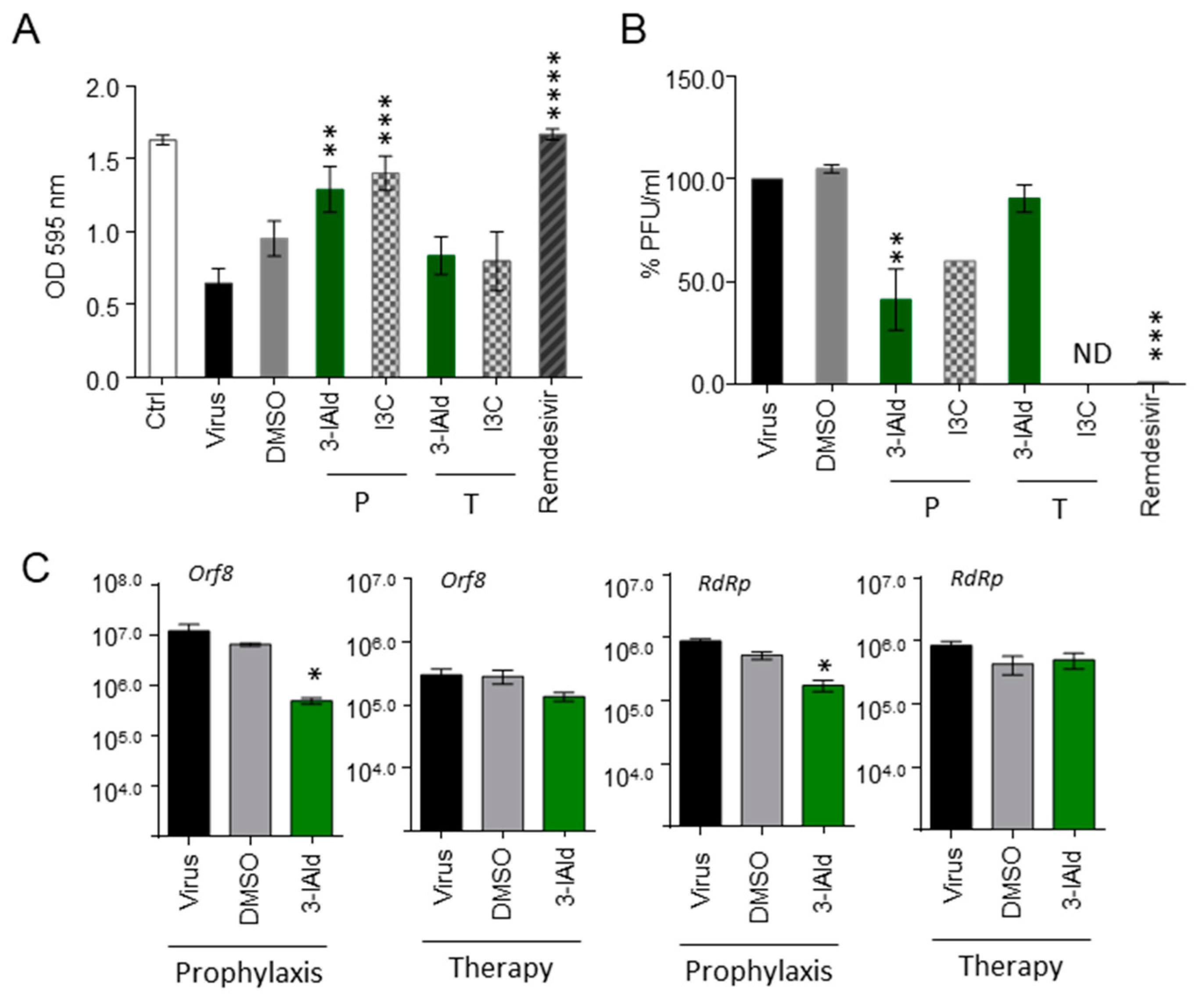
3.5. 3-IAld Exerts Direct Antiviral Effects In Vitro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. WHO Fungal Priority Pathogens List to Guide Research, Development and Public Health Action; 9240060251; Organización Mundial de la Salud (OMS): Geneva, Switzerland, 2022. [Google Scholar]
- Denning, D.W. Global incidence and mortality of severe fungal disease. Lancet Infect. Dis. 2024, 24, e428–e438. [Google Scholar] [CrossRef] [PubMed]
- van de Veerdonk, F.L.; Gresnigt, M.S.; Romani, L.; Netea, M.G.; Latge, J.P. Aspergillus fumigatus morphology and dynamic host interactions. Nat. Rev. Microbiol. 2017, 15, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Costantini, C.; van de Veerdonk, F.L.; Romani, L. COVID-19-Associated Pulmonary Aspergillosis: The Other Side of the Coin. Vaccines 2020, 8, 713. [Google Scholar] [CrossRef] [PubMed]
- Koehler, P.; Bassetti, M.; Chakrabarti, A.; Chen, S.C.A.; Colombo, A.L.; Hoenigl, M.; Klimko, N.; Lass-Florl, C.; Oladele, R.O.; Vinh, D.C.; et al. Defining and managing COVID-19-associated pulmonary aspergillosis: The 2020 ECMM/ISHAM consensus criteria for research and clinical guidance. Lancet Infect Dis. 2021, 21, e149–e162. [Google Scholar] [CrossRef] [PubMed]
- Muthu, V.; Agarwal, R.; Rudramurthy, S.M.; Thangaraju, D.; Shevkani, M.R.; Patel, A.K.; Shastri, P.S.; Tayade, A.; Bhandari, S.; Gella, V.; et al. Prevalence of co-existent COVID-19-associated pulmonary aspergillosis (CAPA) and its impact on early mortality in patients with COVID-19-associated pulmonary mucormycosis (CAPM). Mycoses 2024, 67, e13745. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, S.M.; Pereira, I.; Feys, S.; Cunha, C.; Chamilos, G.; Hoenigl, M.; Wauters, J.; van de Veerdonk, F.L.; Carvalho, A. Integrating genetic and immune factors to uncover pathogenetic mechanisms of viral-associated pulmonary aspergillosis. mBio 2024, 15, e0198223. [Google Scholar] [CrossRef] [PubMed]
- Gangneux, J.P.; Dannaoui, E.; Fekkar, A.; Luyt, C.E.; Botterel, F.; De Prost, N.; Tadie, J.M.; Reizine, F.; Houze, S.; Timsit, J.F.; et al. Fungal infections in mechanically ventilated patients with COVID-19 during the first wave: The French multicentre MYCOVID study. Lancet Respir. Med. 2022, 10, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Bay, P.; Audureau, E.; Preau, S.; Favory, R.; Guigon, A.; Heming, N.; Gault, E.; Pham, T.; Chaghouri, A.; Turpin, M.; et al. COVID-19 associated pulmonary aspergillosis in critically-ill patients: A prospective multicenter study in the era of Delta and Omicron variants. Ann. Intensive Care 2024, 14, 65. [Google Scholar] [CrossRef]
- Costantini, C. Chapter 15—The immune system and the microbiota: The two sides of mucosal tolerance. In Translational Autoimmunity; Rezaei, N., Ed.; Academic Press: Cambridge, MA, USA, 2022; Volume 1, pp. 297–315. [Google Scholar]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef]
- Puccetti, M.; Pariano, M.; Renga, G.; Santarelli, I.; D’Onofrio, F.; Bellet, M.M.; Stincardini, C.; Bartoli, A.; Costantini, C.; Romani, L.; et al. Targeted Drug Delivery Technologies Potentiate the Overall Therapeutic Efficacy of an Indole Derivative in a Mouse Cystic Fibrosis Setting. Cells 2021, 10, 1601. [Google Scholar] [CrossRef]
- Zelante, T.; Puccetti, M.; Giovagnoli, S.; Romani, L. Regulation of host physiology and immunity by microbial indole-3-aldehyde. Curr. Opin. Immunol. 2021, 70, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Zhao, S.; Jin, G.; Song, M.; Zhi, Y.; Zhao, R.; Ma, F.; Zheng, Y.; Wang, K.; Liu, H.; et al. Cytokine Signature Induced by SARS-CoV-2 Spike Protein in a Mouse Model. Front. Immunol. 2020, 11, 621441. [Google Scholar] [CrossRef] [PubMed]
- Puccetti, M.; Giovagnoli, S.; Zelante, T.; Romani, L.; Ricci, M. Development of Novel Indole-3-Aldehyde-Loaded Gastro-Resistant Spray-Dried Microparticles for Postbiotic Small Intestine Local Delivery. J. Pharm. Sci. 2018, 107, 2341–2353. [Google Scholar] [CrossRef] [PubMed]
- Centofanti, F.; Buono, A.; Verboni, M.; Tomino, C.; Lucarini, S.; Duranti, A.; Pandolfi, P.P.; Novelli, G. Synthetic Methodologies and Therapeutic Potential of Indole-3-Carbinol (I3C) and Its Derivatives. Pharmaceuticals 2023, 16, 240. [Google Scholar] [CrossRef] [PubMed]
- Gidari, A.; Sabbatini, S.; Bastianelli, S.; Pierucci, S.; Busti, C.; Bartolini, D.; Stabile, A.M.; Monari, C.; Galli, F.; Rende, M.; et al. SARS-CoV-2 Survival on Surfaces and the Effect of UV-C Light. Viruses 2021, 13, 408. [Google Scholar] [CrossRef]
- Gidari, A.; Sabbatini, S.; Schiaroli, E.; Bastianelli, S.; Pierucci, S.; Busti, C.; Saraca, L.M.; Capogrossi, L.; Pasticci, M.B.; Francisci, D. Synergistic Activity of Remdesivir-Nirmatrelvir Combination on a SARS-CoV-2 In Vitro Model and a Case Report. Viruses 2023, 15, 1577. [Google Scholar] [CrossRef] [PubMed]
- Zelante, T.; Paolicelli, G.; Fallarino, F.; Gargaro, M.; Vascelli, G.; De Zuani, M.; Fric, J.; Laznickova, P.; Kohoutkova, M.H.; Macchiarulo, A.; et al. A microbially produced AhR ligand promotes a Tph1-driven tolerogenic program in multiple sclerosis. Sci. Rep. 2024, 14, 6651. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- Gargaro, M.; Scalisi, G.; Manni, G.; Mondanelli, G.; Grohmann, U.; Fallarino, F. The Landscape of AhR Regulators and Coregulators to Fine-Tune AhR Functions. Int. J. Mol. Sci. 2021, 22, 757. [Google Scholar] [CrossRef]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H., 3rd; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446.e414. [Google Scholar] [CrossRef]
- Di Stadio, A.; Costantini, C.; Renga, G.; Pariano, M.; Ricci, G.; Romani, L. The Microbiota/Host Immune System Interaction in the Nose to Protect from COVID-19. Life 2020, 10, 345. [Google Scholar] [CrossRef] [PubMed]
- Pennarossa, G.; Arcuri, S.; Pasquariello, R.; Gandolfi, F.; Maranesi, M.; Brevini, T.A.L. Cruciferous vegetable-derived indole-3-carbinol prevents coronavirus cell egression mechanisms in tracheal and intestinal 3D in vitro models. Phytochemistry 2023, 212, 113713. [Google Scholar] [CrossRef] [PubMed]
- Centofanti, F.; Alonzi, T.; Latini, A.; Spitalieri, P.; Murdocca, M.; Chen, X.; Cui, W.; Shang, Q.; Goletti, D.; Shi, Y.; et al. Indole-3-carbinol in vitro antiviral activity against SARS-Cov-2 virus and in vivo toxicity. Cell Death Discov. 2022, 8, 491. [Google Scholar] [CrossRef] [PubMed]
- Novelli, G.; Liu, J.; Biancolella, M.; Alonzi, T.; Novelli, A.; Patten, J.J.; Cocciadiferro, D.; Agolini, E.; Colona, V.L.; Rizzacasa, B.; et al. Inhibition of HECT E3 ligases as potential therapy for COVID-19. Cell Death Dis. 2021, 12, 310. [Google Scholar] [CrossRef] [PubMed]
- Dorababu, A. Indole—A promising pharmacophore in recent antiviral drug discovery. RSC Med. Chem. 2020, 11, 1335–1353. [Google Scholar] [CrossRef] [PubMed]
- Hattori, S.I.; Higashi-Kuwata, N.; Hayashi, H.; Allu, S.R.; Raghavaiah, J.; Bulut, H.; Das, D.; Anson, B.J.; Lendy, E.K.; Takamatsu, Y.; et al. A small molecule compound with an indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2021, 12, 668. [Google Scholar] [CrossRef] [PubMed]
- Boriskin, Y.S.; Leneva, I.A.; Pecheur, E.I.; Polyak, S.J. Arbidol: A broad-spectrum antiviral compound that blocks viral fusion. Curr. Med. Chem. 2008, 15, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Blaising, J.; Levy, P.L.; Polyak, S.J.; Stanifer, M.; Boulant, S.; Pecheur, E.I. Arbidol inhibits viral entry by interfering with clathrin-dependent trafficking. Antivir. Res. 2013, 100, 215–219. [Google Scholar] [CrossRef]
- Blaising, J.; Polyak, S.J.; Pecheur, E.I. Arbidol as a broad-spectrum antiviral: An update. Antivir. Res. 2014, 107, 84–94. [Google Scholar] [CrossRef]
- Zhao, X.; Zhao, L.; Zhao, Y.; Huang, K.; Gong, W.; Yang, Y.; Zhao, L.; Xia, X.; Li, Z.; Sheng, F.; et al. 3-Indoleacetonitrile Is Highly Effective in Treating Influenza A Virus Infection In Vitro and In Vivo. Viruses 2021, 13, 1433. [Google Scholar] [CrossRef]
- Nojomi, M.; Yassin, Z.; Keyvani, H.; Makiani, M.J.; Roham, M.; Laali, A.; Dehghan, N.; Navaei, M.; Ranjbar, M. Effect of Arbidol (Umifenovir) on COVID-19: A randomized controlled trial. BMC Infect. Dis. 2020, 20, 954. [Google Scholar] [CrossRef] [PubMed]
- Jie, X.; Hongmei, Y.; Ping, F.; Kuikui, Z.; Bohan, Y.; Rui, M. Beneficial effect of Arbidol in the management of COVID-19 infection. Aging 2021, 13, 9253–9264. [Google Scholar] [CrossRef]
- Tanimoto, K.; Hirota, K.; Fukazawa, T.; Matsuo, Y.; Nomura, T.; Tanuza, N.; Hirohashi, N.; Bono, H.; Sakaguchi, T. Inhibiting SARS-CoV-2 infection in vitro by suppressing its receptor, angiotensin-converting enzyme 2, via aryl-hydrocarbon receptor signal. Sci. Rep. 2021, 11, 16629. [Google Scholar] [CrossRef]
- Johnson-Weaver, B.T.; Choi, H.W.; Yang, H.; Granek, J.A.; Chan, C.; Abraham, S.N.; Staats, H.F. Nasal Immunization With Small Molecule Mast Cell Activators Enhance Immunity to Co-Administered Subunit Immunogens. Front. Immunol. 2021, 12, 730346. [Google Scholar] [CrossRef]
- Renga, G.; Nunzi, E.; Pariano, M.; Puccetti, M.; Bellet, M.M.; Pieraccini, G.; D’Onofrio, F.; Santarelli, I.; Stincardini, C.; Aversa, F.; et al. Optimizing therapeutic outcomes of immune checkpoint blockade by a microbial tryptophan metabolite. J. Immunother. Cancer 2022, 10, e003725. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Offringa, A.K.; van Eijk, L.E.; Abdulle, A.E.; Hillebrands, J.L.; van der Voort, P.H.J.; van Goor, H.; van Hezik, E.J. N-Acetylcysteine and Hydrogen Sulfide in Coronavirus Disease 2019. Antioxid. Redox Signal. 2021, 35, 1207–1225. [Google Scholar] [CrossRef]
- Liu, Y.; Lv, J.; Liu, J.; Li, M.; Xie, J.; Lv, Q.; Deng, W.; Zhou, N.; Zhou, Y.; Song, J.; et al. Mucus production stimulated by IFN-AhR signaling triggers hypoxia of COVID-19. Cell Res. 2020, 30, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, F.; Li, Z.; Remes-Lenicov, F.; Davola, M.E.; Elizalde, M.; Paletta, A.; Ashkar, A.A.; Mossman, K.L.; Dugour, A.V.; Figueroa, J.M.; et al. AHR signaling is induced by infection with coronaviruses. Nat. Commun. 2021, 12, 5148. [Google Scholar] [CrossRef]
- Shi, J.; Du, T.; Wang, J.; Tang, C.; Lei, M.; Yu, W.; Yang, Y.; Ma, Y.; Huang, P.; Chen, H.; et al. Aryl hydrocarbon receptor is a proviral host factor and a candidate pan-SARS-CoV-2 therapeutic target. Sci. Adv. 2023, 9, eadf0211. [Google Scholar] [CrossRef] [PubMed]
- Haid, S.; Matthaei, A.; Winkler, M.; Sake, S.M.; Gunesch, A.P.; Milke, V.; Kohler, N.M.; Ruckert, J.; Vieyres, G.; Kuhl, D.; et al. Repurposing screen identifies novel candidates for broad-spectrum coronavirus antivirals and druggable host targets. Antimicrob. Agents Chemother. 2024, 68, e0121023. [Google Scholar] [CrossRef]
- Boule, L.A.; Burke, C.G.; Jin, G.B.; Lawrence, B.P. Aryl hydrocarbon receptor signaling modulates antiviral immune responses: Ligand metabolism rather than chemical source is the stronger predictor of outcome. Sci. Rep. 2018, 8, 1826. [Google Scholar] [CrossRef] [PubMed]
- Holloman, B.L.; Cannon, A.; Wilson, K.; Nagarkatti, P.; Nagarkatti, M. Aryl Hydrocarbon Receptor Activation Ameliorates Acute Respiratory Distress Syndrome through Regulation of Th17 and Th22 Cells in the Lungs. mBio 2023, 14, e0313722. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Faber, S.C. And Now for Something Completely Different: Diversity in Ligand-Dependent Activation of Ah Receptor Responses. Curr. Opin. Toxicol. 2017, 2, 124–131. [Google Scholar] [CrossRef]
- Swimm, A.; Giver, C.R.; DeFilipp, Z.; Rangaraju, S.; Sharma, A.; Ulezko Antonova, A.; Sonowal, R.; Capaldo, C.; Powell, D.; Qayed, M.; et al. Indoles derived from intestinal microbiota act via type I interferon signaling to limit graft-versus-host disease. Blood 2018, 132, 2506–2519. [Google Scholar] [CrossRef]
- Deprez, M.; Zaragosi, L.E.; Truchi, M.; Becavin, C.; Ruiz Garcia, S.; Arguel, M.J.; Plaisant, M.; Magnone, V.; Lebrigand, K.; Abelanet, S.; et al. A Single-Cell Atlas of the Human Healthy Airways. Am. J. Respir. Crit. Care Med. 2020, 202, 1636–1645. [Google Scholar] [CrossRef]
- Vieira Braga, F.A.; Kar, G.; Berg, M.; Carpaij, O.A.; Polanski, K.; Simon, L.M.; Brouwer, S.; Gomes, T.; Hesse, L.; Jiang, J.; et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 2019, 25, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef]
- Costantini, C.; Nunzi, E.; Spolzino, A.; Palmieri, M.; Renga, G.; Zelante, T.; Englmaier, L.; Coufalikova, K.; Spacil, Z.; Borghi, M.; et al. Pharyngeal Microbial Signatures Are Predictive of the Risk of Fungal Pneumonia in Hematologic Patients. Infect. Immun. 2021, 89, e0010521. [Google Scholar] [CrossRef]
- Costantini, C.; Nunzi, E.; Spolzino, A.; Merli, F.; Facchini, L.; Spadea, A.; Melillo, L.; Codeluppi, K.; Marchesi, F.; Marchesini, G.; et al. A High-Risk Profile for Invasive Fungal Infections Is Associated with Altered Nasal Microbiota and Niche Determinants. Infect. Immun. 2022, 90, e0004822. [Google Scholar] [CrossRef]
- Costantini, C.; Nunzi, E.; Romani, L. From the nose to the lungs: The intricate journey of airborne pathogens amid commensal bacteria. Am. J. Physiol. Cell Physiol. 2022, 323, C1036–C1043. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Murine Primers | |
| β-actin (Beta-Actin) | forward AGCCATGTACGTAGCCATCC reverse CTCTCAGCTGTGGTGGTGAA |
| Ace2 (Angiotensin-Converting Enzyme 2) | forward TCCATT-GGTCTTCTGCCATCC reverse AACGATCTCCCGCTTCATCTC |
| Ahr (Aryl Hydrocarbon Receptor) | forward TCCATCCTGGAAATTCGAACC reverse TCTTCATCCGTCAGTGGTCTC |
| Ahrr (Aryl Hydrocarbon Receptor Repressor) | forward AGAGGGTTCCCCGTGCAG reverse ACTCACCACCAGAGCGAAGC |
| Cyp1b1 (Cytochrome P450 Family 1 Subfam.B Member 1) | forward TTCTCCAGCTTTTTGCCTGT reverse TAATGAAGCCGTCCTTGTCC |
| Human Primers | |
| β-actin (Beta-Actin) | forward CACTCTTCCAGCCTTCCTTCC reverse ACAGCACTGTGTTGGCGTAC |
| IL1B (Interleukin 1 Beta) | forward AAGCTCCTGTGGCAATTGAA reverse TCCTCCTTCTGGAACTGCTG |
| IL10 (Interleukin 10) | forward CCTGCCTAACATGCTTCGAGA reverse TCTTGGTTCTCAGCTTGGGG |
| IFNA1 (Interferon Alpha 1) | forward ACCCACAGCCTGGATAACAG reverse ACTGGTTGCCATCAAACTCC |
| IFNB1 (Interferon Beta 1) | forward AGTAGGCGACACTGTTCGTG reverse GCCTCCCATTCAATTGCCAC |
| OAS1 (2′-5′-Oligoadenylate Synthetase 1) | forward GAGACCCAAAGGGTTGGAGG reverse TCATCGTCTGCACTGTTGCT |
| PPARG (Peroxisome Proliferator-Activated Receptor Gamma) | forward TCGAGGACACCGGAGAGG reverse CACGGAGCTGATCCCAAAGT |
| NQO1 (NAD(P)H Quinone Dehydrogenase 1) | forward GGTTTGGAGTCCCTGCCATT reverse ACCAGTGGTGATGGAAAGCA |
| HMOX1 (Heme Oxygenase 1) | forward TGACCCATGACACCAAGGAC reverse AGTGTAAGGACCCATCGGAGA. |
| Viral Primers | |
| Orf8 (Open Reading Frame 8) | forward GGTGCTGACTGAGAGCAATAA reverse CACATTAGAGCCGGTTGAGTAG |
| RdRp (RNA-dependent RNA polymerase) | Forward ACGCTCAAAGCTACTGAGGAGAC reverse GGTCTAGGTTTACCAACTTCCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pariano, M.; Gidari, A.; Stincardini, C.; Pierucci, S.; Bastianelli, S.; Puccetti, M.; Giovagnoli, S.; Bellet, M.M.; Fabi, C.; Castronari, R.; et al. Protective Effect of Indole-3-Aldehyde in Murine COVID-19-Associated Pulmonary Aspergillosis. J. Fungi 2024, 10, 510. https://doi.org/10.3390/jof10070510
Pariano M, Gidari A, Stincardini C, Pierucci S, Bastianelli S, Puccetti M, Giovagnoli S, Bellet MM, Fabi C, Castronari R, et al. Protective Effect of Indole-3-Aldehyde in Murine COVID-19-Associated Pulmonary Aspergillosis. Journal of Fungi. 2024; 10(7):510. https://doi.org/10.3390/jof10070510
Chicago/Turabian StylePariano, Marilena, Anna Gidari, Claudia Stincardini, Sara Pierucci, Sabrina Bastianelli, Matteo Puccetti, Stefano Giovagnoli, Marina M. Bellet, Consuelo Fabi, Roberto Castronari, and et al. 2024. "Protective Effect of Indole-3-Aldehyde in Murine COVID-19-Associated Pulmonary Aspergillosis" Journal of Fungi 10, no. 7: 510. https://doi.org/10.3390/jof10070510