
Genomic Sequencing and Functional Analysis of the Ex-Type Strain of Malbranchea zuffiana

 ,
,  and
and 
Abstract
:1. Introduction
2. Material and Methods
2.1. DNA Extraction for Genome Sequencing
2.2. Genome Assembly and Annotation
2.3. Phylogenetic and Functional Comparative Analysis
3. Results and Discussion
3.1. Genome Assembly and Evaluation
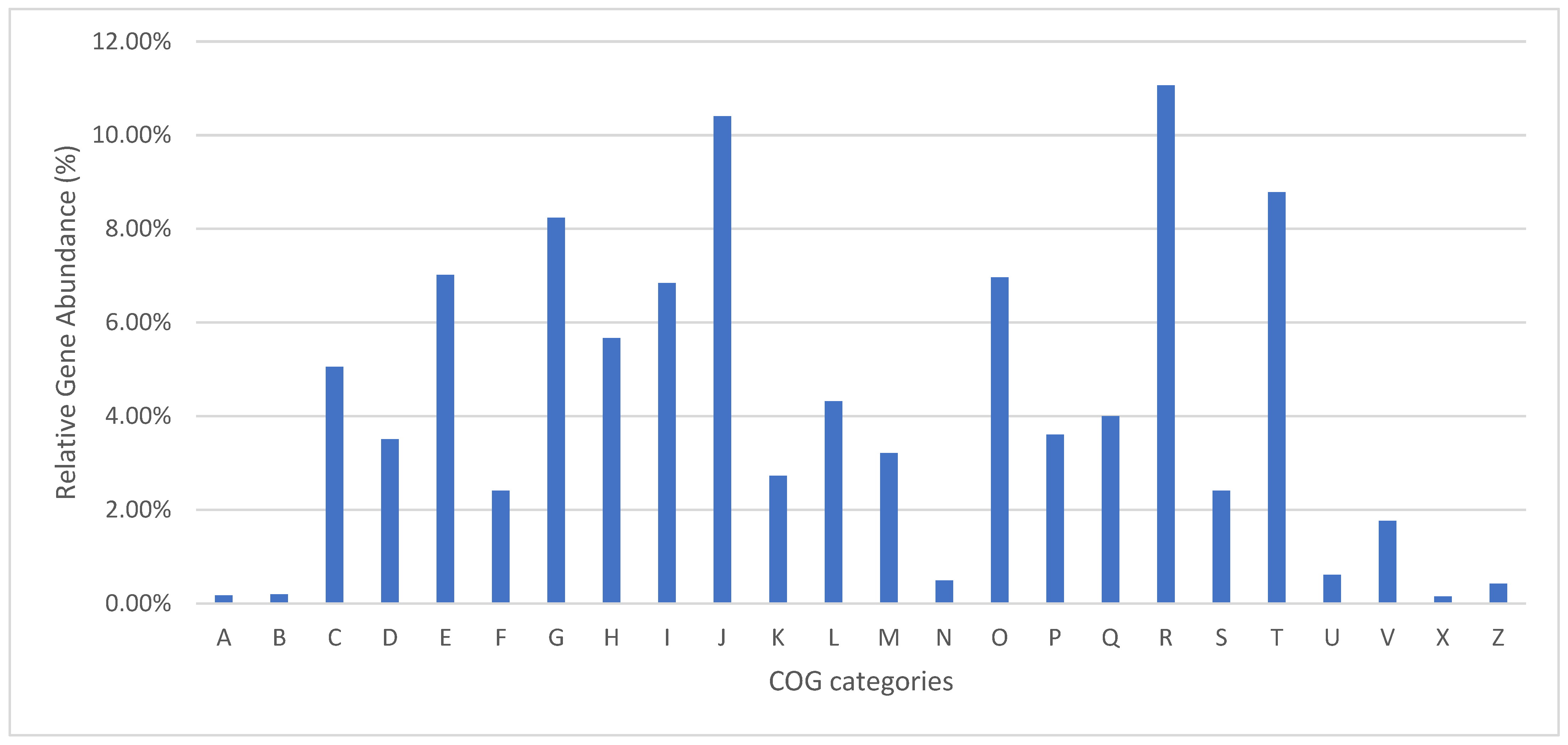
3.2. Genome Annotation
3.3. AntiSMASH
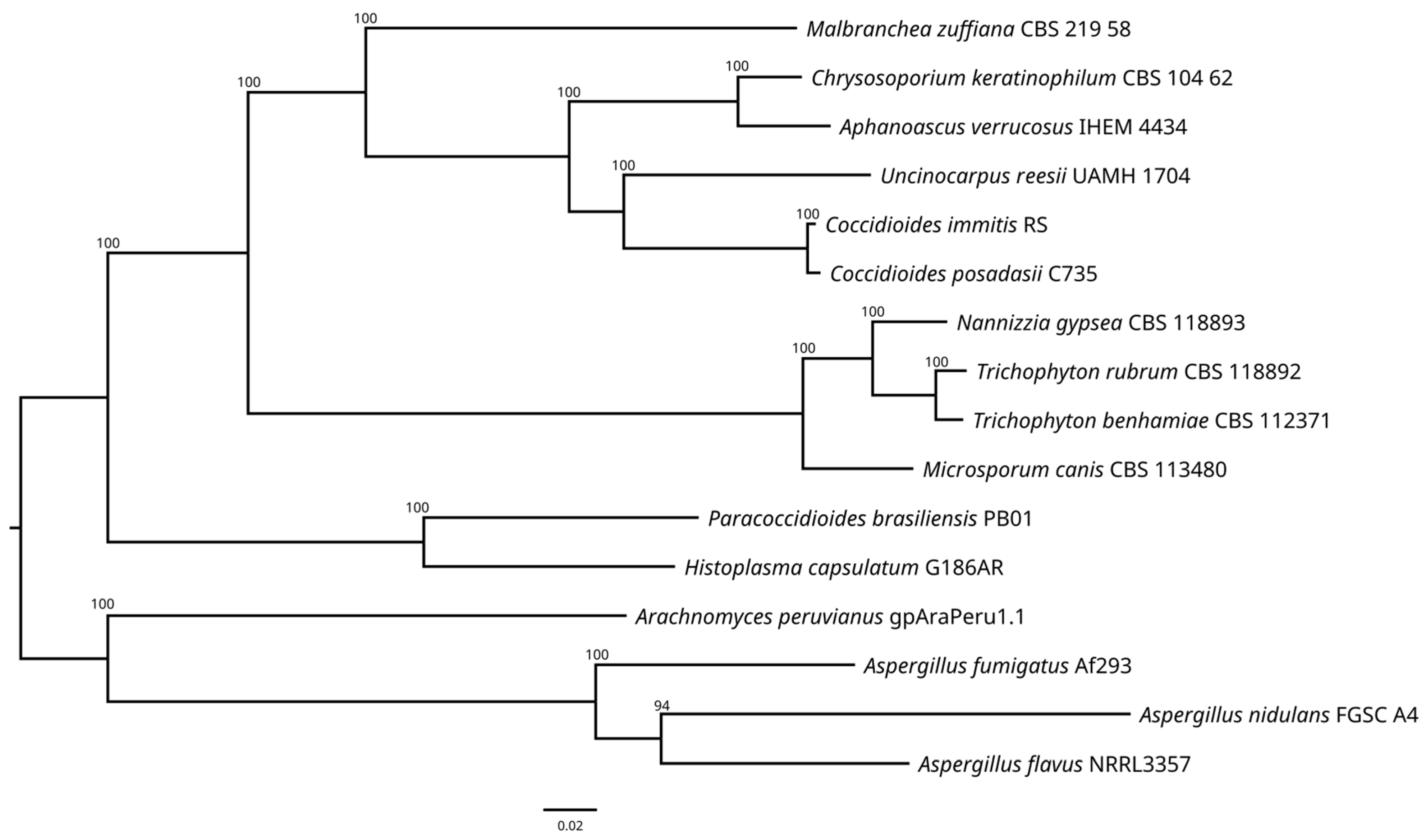
3.4. Phylogenomics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saccardo, P. Fungi Gallici lecti a Cl. viris P. Brunaud, C.C. Gillet, Abb. Letendre, A. Malbranche, J. Therry & Dom. Libert. Ser. IV. Michelia 1882, 2, 583–648. [Google Scholar]
- Cooney, D.G.; Emerson, R. Thermophilic Fungi: An Account of Their Biology; Freeman, W.H., Ed.; Activities and Classification: San Francisco, CA, USA; London, UK, 1964. [Google Scholar]
- Sigler, L.; Carmichael, J. Taxonomy of Malbranchea and some other hyphomycetes with arthroconidia. Mycotaxon 1976, 4, 349–488. [Google Scholar]
- Sigler, L.; Hambleton, S.; Flis, A.L.; Paré, J.A. Auxarthron teleomorphs for Malbranchea filamentosa and Malbranchea albolutea and relationships within Auxarthron. Stud. Mycol. 2002, 47, 111–122. [Google Scholar]
- Rodríguez-Andrade, E.; Cano-Lira, J.F.; Wiederhold, N.; Pérez-Cantero, A.; Guarro, J.; Stchigel, A.M. A revision of malbranchea-like fungi from clinical specimens in the United States of America reveals unexpected novelty. IMA Fungus 2021, 12, 25. [Google Scholar] [CrossRef]
- Kandemir, H.; Dukik, K.; de Melo Teixeira, M.; Stielow, J.B.; Delma, F.Z.; Al-Hatmi, A.M.S.; Ahmed, S.A.; Ilkit, M.; de Hoog, G.S. Phylogenetic and ecological reevaluation of the order Onygenales. Fungal Divers. 2022, 115, 1–72. [Google Scholar] [CrossRef]
- Torres-Garcia, D.; Gené, J.; García, D.; Cano-Lira, J.F. Insights into Some Onygenalean Fungi from Freshwater Sediments in Spain and Description of Novel Taxa. J. Fungi 2023, 9, 1129. [Google Scholar] [CrossRef]
- Gibas, C.F.C.; Sigler, L.; Summerbell, R.C.; Currah, R.S. Phylogeny of the genus Arachnomyces and its anamorphs and the establishment of Arachnomycetales, a new eurotiomycete order in the Ascomycota. Stud. Mycol. 2002, 47, 131–139. [Google Scholar]
- Guarro, J.; Punsola, L.; Calvo, M.A. Keratinophilic fungi from soil of Tarragona, Catalunya. Mycopathologia 1981, 76, 69–71. [Google Scholar] [CrossRef]
- Deshmukh, S.K.; Verekar, S.A.; Chavan, Y.G. Keratinophilic fungi from the vicinity of salt pan soils of Sambhar lake Rajasthan (India). J. Mycol. Med. 2018, 28, 457–461. [Google Scholar] [CrossRef]
- Pan, W.Z.; Huang, X.W.; Wei, K.B.; Zhang, C.M.; Yang, D.M.; Ding, J.M.; Zhang, K.Q. Diversity of thermophilic fungi in Tengchong Rehai national park revealed by ITS nucleotide sequence analyses. J. Microbiol. 2010, 48, 146–152. [Google Scholar] [CrossRef]
- Pakshir, K.; Rahimi Ghiasi, M.; Zomorodian, K.; Gharavi, A.R. Isolation and molecular identification of keratinophilic fungi from public parks soil in Shiraz, Iran. Biomed Res. Int. 2013, 2013, 619576. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, S.K. Keratinophilic fungi on feathers of pigeon in Maharashtra, India. Mycoses 2004, 47, 213–215. [Google Scholar] [CrossRef]
- Lionakis, M.S.; Kontoyiannis, D.P. The significance of isolation of saprophytic molds from the lower respiratory tract in patients with cancer. Cancer 2004, 100, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, R.F.; Pyun, J.; Modena, B.D.; Crum-Cianflone, N. A rare case of Malbranchea species pulmonary infection in the setting of suspected selective immunoglobulin M deficiency. Infect. Dis. Clin. Pract. 2019, 27, 47–51. [Google Scholar] [CrossRef]
- Bamahfouz, A.Y.; Alsaidi, A.A.; Alharbi, I.J.; Elsebaei, E.A.; Aldosari, A.M.; Farahat, A.G.; Alhazmi, R.T. Indolent keratitis due to fungus of Malbranchea species. A case report. Ann. Med. Surg. 2020, 60, 606–609. [Google Scholar] [CrossRef]
- Stein, M.; Tran, V.; Nichol, K.A.; Lagacé-Wiens, P.; Pieroni, P.; Adam, H.J.; Turenne, C.; Walkty, A.J.; Normand, A.C.; Hendrickx, M.; et al. Evaluation of three MALDI-TOF mass spectrometry libraries for the identification of filamentous fungi in three clinical microbiology laboratories in Manitoba, Canada. Mycoses 2018, 61, 743–753. [Google Scholar] [CrossRef] [PubMed]
- McCormick-Baw, C.; Clark, A.; Knights, S.; Perl, T.; Cavuoti, D. The Brief Case: Encephalitis in a West Texas Woman. J. Clin. Microbiol. 2022, 60, e0227121. [Google Scholar] [CrossRef]
- Borman, A.M.; Johnson, E.M. Name Changes for Fungi of Medical Importance, 2020 to 2021. J. Clin. Microbiol. 2023, 61, e0033022. [Google Scholar] [CrossRef]
- Hubka, V.; Dobiasova, S.; Lyskova, P.; Mallatova, N.; Chlebkova, J.; Skorepova, M.; Kubatova, A.; Dobias, R.; Chudickova, M.; Kolarik, M. Auxarthron ostraviense sp. nov., and A. umbrinum associated with non-dermatophytic onychomycosis. Med. Mycol. 2013, 51, 614–624. [Google Scholar] [CrossRef]
- Gharib, S.J.; Abdullah, S.K.; Richardson, M.D. Auxarthron alboluteum related to non-dermatophytic toenail infection in Kurdistan region, Iraq: A case report. Med. Mycol. Case Rep. 2019, 26, 53–56. [Google Scholar] [CrossRef]
- Tang, S.; Prem, A.; Tjokrosurjo, J.; Sary, M.; Van Bel, M.A.; Rodrigues-Hoffmann, A.; Kavanagh, M.; Wu, G.; Van Eden, M.E.; Krumbeck, J.A. The canine skin and ear microbiome: A comprehensive survey of pathogens implicated in canine skin and ear infections using a novel next-generation-sequencing-based assay. Vet. Microbiol. 2020, 247, 108764. [Google Scholar] [CrossRef] [PubMed]
- Ong, P.S.; Gaucher, G.M. Production, purification and partial characterization of an extracellular protease from a thermophilic fungus. In Proceedings of the IVth International Fermentation Symposium, Kyoto, Japan, 19–25 March 1972; Terui, G., Ed.; Society of Fermentation Technology: Osaka, Japan, 1972; pp. 271–278. [Google Scholar]
- Martínez-Luis, S.; Rodríguez, R.; Acevedo, L.; González, M.C.; Lira-Rocha, A.; Mata, R. Malbrancheamide, a new calmodulin inhibitor from the fungus Malbranchea aurantiaca. Tetrahedron 2006, 62, 1817–1822. [Google Scholar] [CrossRef]
- Figueroa, M.; González, M.D.C.; Mata, R. Malbrancheamide B, a novel compound from the fungus Malbranchea aurantiaca. Nat. Prod. Res. 2008, 22, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Luis, S.; González, M.C.; Ulloa, M.; Mata, R. Phytotoxins from the fungus Malbranchea aurantiaca. Phytochemistry 2005, 66, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Greshock, T.J.; Miller, K.A.; Sherman, D.H.; Williams, R.M. Premalbrancheamide: Synthesis, isotopic labeling, biosynthetic incorporation, and detection in cultures of Malbranchea aurantiaca. Org. Lett. 2008, 10, 4863–4866. [Google Scholar] [CrossRef] [PubMed]
- Rebollar-Ramos, D.; Ovalle-Magallanes, B.; Palacios-Espinosa, J.F.; Macías-Rubalcava, M.L.; Raja, H.A.; González-Andrade, M.; Mata, R. α-Glucosidase and PTP-1B Inhibitors from Malbranchea dendritica. ACS Omega 2021, 6, 22969–22981. [Google Scholar] [CrossRef]
- Wakana, D.; Itabashi, T.; Kawai, K.I.; Yaguchi, T.; Fukushima, K.; Goda, Y.; Hosoe, T. Cytotoxic anthrasteroid glycosides, malsterosides A–C, from Malbranchea filamentosa. J. Antibiot. 2014, 67, 585–588. [Google Scholar] [CrossRef]
- Verastegui-Omaña, B.; Rebollar-Ramos, D.; Pérez-Vásquez, A.; Martínez, A.L.; Madariaga-Mazón, A.; Flores-Bocanegra, L.; Mata, R. α-Glucosidase inhibitors from Malbranchea flavorosea. J. Nat. Prod. 2017, 80, 190–195. [Google Scholar] [CrossRef]
- Watts, K.R.; Loveridge, S.T.; Tenney, K.; Media, J.; Valeriote, F.A.; Crews, P. Utilizing DART mass spectrometry to pinpoint halogenated metabolites from a marine invertebrate-derived fungus. J. Org. Chem. 2011, 76, 6201–6208. [Google Scholar] [CrossRef]
- Monteiro, L.M.O.; Vici, A.C.; Messias, J.M.; Heinen, P.R.; Pinheiro, V.E.; Rechia, C.G.V.; Buckeridge, M.S.; de Moraes, M.D.L.T. Increased Malbranchea pulchella β-glucosidase production and its application in agroindustrial residue hydrolysis: A research based on experimental designs. Biotechnol. Rep. 2021, 30, e00618. [Google Scholar] [CrossRef]
- Saito, M.; Matsuura, I.; Okazaki, H. Tf-26Vx, an antibiotic produced by a thermophilic fungus. J. Antibiot. 1979, 32, 1210–1212. [Google Scholar] [CrossRef]
- Matsuo, M.; Yasui, T.; Kobayashi, T. Purification and Some Properties of β-Xylosidase from Malbranchea pulchella var. sulfurea No. 48. Agric. Biol. Chem. 1977, 41, 1593–1599. [Google Scholar] [CrossRef]
- Matsuo, M.; Yasui, T.; Kobayashi, T. Enzymatic Properties of β-Xylosidase from Malbranchea pulchella var. sulfurea No. 48. Agric. Biol. Chem. 2014, 41, 1601–1606. [Google Scholar] [CrossRef]
- McKay, D.J.; Stevenson, K.J. Lipoamide Dehydrogenase from Malbranchea pulchella: Isolation and Characterization. Biochemistry 1979, 18, 4702–4707. [Google Scholar] [CrossRef]
- Ong, P.S.; Gaucher, G.M. Protease production by thermophilic fungi. Can. J. Microbiol. 1973, 19, 129–133. [Google Scholar] [CrossRef]
- Voordouw, G.; Gaucher, G.M.; Roche, R.S. Physiochemical properties of thermomycolase, the thermostable, extracellular, serine protease of the fungus Malbranchea pulchella. Can. J. Biochem. 1974, 52, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Ong, P.S.; Gaucher, G.M. Production, purification and characterization of thermomycolase, the extracellular serine protease of the thermophilic fungus Malbranchea pulchella var. sulfurea. Can. J. Microbiol. 1976, 22, 165–176. [Google Scholar] [CrossRef]
- Pereira, M.G.; Guimarães, L.H.S.; Furriel, R.P.M.; Polizeli, M.d.L.T.d.M.; Terenzi, H.F.; Jorge, J.A. Biochemical properties of an extracellular trehalase from Malbranchea pulchella var. sulfurea. J. Microbiol. 2011, 49, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, M.; Yasui, T.; Kobayashi, T. Production and saccharifying action for xylan of xylanase from Malbranchea pulchella var. sulfurea no. 48. Nippon. Nagaikagaku Kaishi 1975, 49, 263–270. [Google Scholar] [CrossRef]
- Matsuo, M.; Yasui, T. Properties of Xylanase of Malbranchea pulchella var. sulfurea. Agric. Biol. Chem. 1985, 49, 839–841. [Google Scholar] [CrossRef]
- Andrews, S. FastQC A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 8 July 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Zimin, A.V.; Puiu, D.; Luo, M.-C.; Zhu, T.; Koren, S.; Marçais, G.; Yorke, J.A.; Dvořák, J.; Salzberg, S.L. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 2017, 27, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Sim, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M.; Rosenberg, M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Seemann, T. Barrnap. Available online: https://github.com/tseemann/barrnap (accessed on 8 July 2022).
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. In Gene Prediction; Methods in Molecular Biology; Humana: New York, NY, USA, 2019; Volume 1962, pp. 1–14. [Google Scholar] [CrossRef]
- Brůna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics Bioinforma. 2021, 3, lqaa108. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; De Silva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. PHI-base: The pathogen–host interactions database. Nucleic Acids Res. 2020, 48, D613–D620. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 466201. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Darriba, D.i.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Diep, A.L.; Hoyer, K.K. Host Response to Coccidioides Infection: Fungal Immunity. Front. Cell. Infect. Microbiol. 2020, 10, 581101. [Google Scholar] [CrossRef]
- Powell, D.; Jones, A.; Jackson, N.; Kaur, P.; Bar, I.; Schwessinger, B.; Frère, C.H. Genome Sequence of the Fungus Nannizziopsis barbatae, an Emerging Reptile Pathogen. Microbiol. Resour. Announc. 2021, 10, e01213-20. [Google Scholar] [CrossRef]
- Bork, P. Hundreds of ankyrin-like repeats in functionally diverse proteins: Mobile modules that cross phyla horizontally? Proteins Struct. Funct. Bioinform. 1993, 17, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Jain, B.P.; Pandey, S. WD40 Repeat Proteins: Signalling Scaffold with Diverse Functions. Protein J. 2018, 37, 391–406. [Google Scholar] [CrossRef]
- Pang, K.; Wang, W.; Qin, J.X.; Shi, Z.D.; Hao, L.; Ma, Y.Y.; Xu, H.; Wu, Z.X.; Pan, D.; Chen, Z.S.; et al. Role of protein phosphorylation in cell signaling, disease, and the intervention therapy. MedComm 2022, 3, e175. [Google Scholar] [CrossRef] [PubMed]
- Ten years of CAZypedia: A living encyclopedia of carbohydrate-active enzymes. Glycobiology 2018, 28, 3–8. [CrossRef]
- Balabanova, L.; Slepchenko, L.; Son, O.; Tekutyeva, L. Biotechnology potential of marine fungi degrading plant and algae polymeric substrates. Front. Microbiol. 2018, 9, 374409. [Google Scholar] [CrossRef]
- Orłowska, M.; Muszewska, A. In Silico Predictions of Ecological Plasticity Mediated by Protein Family Expansions in Early-Diverging Fungi. J. Fungi 2022, 8, 67. [Google Scholar] [CrossRef]
- Barrett, K.; Jensen, K.; Meyer, A.S.; Frisvad, J.C.; Lange, L. Fungal secretome profile categorization of CAZymes by function and family corresponds to fungal phylogeny and taxonomy: Example Aspergillus and Penicillium. Sci. Rep. 2020, 10, 5158. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, H.; Wang, C.; Xu, J.R. Correction to Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genom. 2014, 14, 274. [Google Scholar] [CrossRef]
- López-Mondéjar, R.; Zühlke, D.; Větrovský, T.; Becher, D.; Riedel, K.; Baldrian, P. Decoding the complete arsenal for cellulose and hemicellulose deconstruction in the highly efficient cellulose decomposer Paenibacillus O199. Biotechnol. Biofuels 2016, 9, 104. [Google Scholar] [CrossRef]
- Fernandez, J.; Lopez, V.; Kinch, L.; Pfeifer, M.A.; Gray, H.; Garcia, N.; Grishin, N.V.; Khang, C.H.; Orth, K. Role of two metacaspases in development and pathogenicity of the rice blast fungus Magnaporthe oryzae. mBio 2021, 12, e03471-20. [Google Scholar] [CrossRef]
- Cowen, L.E.; Singh, S.D.; Köhler, J.R.; Collins, C.; Zaas, A.K.; Schell, W.A.; Aziz, H.; Mylonakis, E.; Perfect, J.R.; Whitesell, L.; et al. Harnessing Hsp90 function as a powerful, broadly effective therapeutic strategy for fungal infectious disease. Proc. Natl. Acad. Sci. USA 2009, 106, 2818–2823. [Google Scholar] [CrossRef]
- Lamoth, F.; Juvvadi, P.R.; Gehrke, C.; Asfaw, Y.G.; Steinbach, W.J. Transcriptional Activation of Heat Shock Protein 90 Mediated Via a Proximal Promoter Region as Trigger of Caspofungin Resistance in Aspergillus fumigatus. J. Infect. Dis. 2014, 209, 473–481. [Google Scholar] [CrossRef]
- Bui, D.C.; Lee, Y.; Lim, J.Y.; Fu, M.; Kim, J.C.; Choi, G.J.; Son, H.; Lee, Y.W. Heat shock protein 90 is required for sexual and asexual development, virulence, and heat shock response in Fusarium graminearum. Sci. Rep. 2016, 6, 28154. [Google Scholar] [CrossRef]
- Crits-Christoph, A.; Bhattacharya, N.; Olm, M.R.; Song, Y.S.; Banfield, J.F. Transporter genes in biosynthetic gene clusters predict metabolite characteristics and siderophore activity. Genome Res. 2021, 31, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Rokas, A.; Mead, M.E.; Steenwyk, J.L.; Raja, H.A.; Oberlies, N.H. Biosynthetic gene clusters and the evolution of fungal chemodiversity. Nat. Prod. Rep. 2020, 37, 868–878. [Google Scholar] [CrossRef]
- Prosperini, A.; Berrada, H.; Ruiz, M.J.; Caloni, F.; Coccini, T.; Spicer, L.J.; Perego, M.C.; Lafranconi, A. A review of the mycotoxin enniatin B. Front. Public Health 2017, 5, 300387. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, V.C.; Mirabelli, V.; Cimmarusti, M.T.; Haidukowski, M.; Leslie, J.F.; Logrieco, A.F.; Caliandro, R.; Fanelli, F.; Mulè, G. Enniatin and Beauvericin Biosynthesis in Fusarium Species: Production Profiles and Structural Determinant Prediction. Toxins 2017, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.J.; Koulman, A.; Christensen, M.; Lane, G.A.; Fraser, K.; Forester, N.; Johnson, R.D.; Bryan, G.T.; Rasmussen, S. An Extracellular Siderophore Is Required to Maintain the Mutualistic Interaction of Epichloë festucae with Lolium perenne. PLoS Pathog. 2013, 9, e1003332. [Google Scholar] [CrossRef] [PubMed]
- Godio, R.P.; Martín, J.F. Modified oxidosqualene cyclases in the formation of bioactive secondary metabolites: Biosynthesis of the antitumor clavaric acid. Fungal Genet. Biol. 2009, 46, 232–242. [Google Scholar] [CrossRef]
- Granados-Casas, A.O.; Sastoque, A.P.; Stchigel, A.M.; Fernández-Bravo, A.; Cano-Lira, J.F. Hybrid De Novo Whole-Genome Assembly, Annotation, and Identification of Secondary Metabolite Gene Clusters in the Ex-Type Strain of Chrysosporium keratinophilum. J. Fungi 2023, 9, 389. [Google Scholar] [CrossRef]
- Hai, Y.; Huang, A.M.; Tang, Y. Structure-guided function discovery of an NRPS-like glycine betaine reductase for choline biosynthesis in fungi. Proc. Natl. Acad. Sci. USA 2019, 116, 10348–10353. [Google Scholar] [CrossRef] [PubMed]
- Markham, P.; Robson, G.D.; Bainbridge, B.W.; Trinci, A.P.J. Choline: Its role in the growth of filamentous fungi and the regulation of mycelial morphology. FEMS Microbiol. Rev. 1993, 10, 287–300. [Google Scholar] [CrossRef]
- Fraley, A.E.; Garcia-Borràs, M.; Tripathi, A.; Khare, D.; Mercado-Marin, E.V.; Tran, H.; Dan, Q.; Webb, G.P.; Watts, K.R.; Crews, P.; et al. Function and structure of MalA/MalA’, iterative halogenases for late-stage C–H functionalization of indole alkaloids. J. Am. Chem. Soc. 2017, 139, 12060. [Google Scholar] [CrossRef]
- Li, S.; Srinivasan, K.; Tran, H.; Yu, F.; Finefield, J.M.; Sunderhaus, J.D.; McAfoos, T.J.; Tsukamoto, S.; Williams, R.M.; Sherman, D.H. Comparative analysis of the biosynthetic systems for fungal bicyclo [2.2.2]diazaoctane indole alkaloids: The (+)/(−)-notoamide, paraherquamide and malbrancheamide pathways. MedChemComm 2012, 3, 987–996. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Han, Y.F.; Chen, W.H.; Liang, Z.Q. Phylogeny and taxonomy of three new Ctenomyces (Arthrodermataceae, Onygenales) species from China. MycoKeys 2019, 47, 1–16. [Google Scholar] [CrossRef]
- Scott, J.A.; Untereiner, W.A. Determination of keratin degradation by fungi using keratin azure. Med. Mycol. 2004, 42, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Rajak, R.C. Keratinophilic fungi: Nature’s keratin degrading machines! Resonance 2003, 8, 28–40. [Google Scholar] [CrossRef]
- Taylor, M.L.; Reyes-Montes, M.d.R.; Estrada-Bárcenas, D.A.; Zancopé-Oliveira, R.M.; Rodríguez-Arellanes, G.; Ramírez, J.A. Considerations about the Geographic Distribution of Histoplasma Species. Appl. Environ. Microbiol. 2022, 88, e0201021. [Google Scholar] [CrossRef] [PubMed]
- Hahn, R.C.; Hagen, F.; Mendes, R.P.; Burger, E.; Nery, A.F.; Siqueira, N.P.; Guevara, A.; Rodrigues, A.M.; de Camargo, Z.P. Paracoccidioidomycosis: Current Status and Future Trends. Clin. Microbiol. Rev. 2022, 35, e0023321. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Johnston, P.R.; Takamatsu, S.; Spatafora, J.W.; Hibbett, D.S. Toward a phylogenetic classification of the Leotiomycetes based on rDNA data. Mycologia 2006, 98, 1065–1075. [Google Scholar] [CrossRef]
- Keller, N.P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [Google Scholar] [CrossRef]
- Avalos, J.; Limón, M.C. Fungal Secondary Metabolism. Encyclopedia 2022, 2, 1–13. [Google Scholar] [CrossRef]
- Devi, R.; Kaur, T.; Guleria, G.; Rana, K.L.; Kour, D.; Yadav, N.; Yadav, A.N.; Saxena, A.K. Fungal secondary metabolites and their biotechnological applications for human health. In New and Future Developments in Microbial Biotechnology and Bioengineering; Elsevier: Amsterdam, The Netherlands, 2020; pp. 147–161. [Google Scholar] [CrossRef]
- Maresca, M.; Muchagato Mauricio, E.; Brito, L.; Conrado, R.; Colombo Gomes, T.; Sales Calaço Roque, G.; Olívia De Souza, A. Overview of Bioactive Fungal Secondary Metabolites: Cytotoxic and Antimicrobial Compounds. Antibiotics 2022, 11, 1604. [Google Scholar] [CrossRef]
- Kramer, R.; Abraham, W.R. Volatile sesquiterpenes from fungi: What are they good for? Phytochem. Rev. 2011, 11, 15–37. [Google Scholar] [CrossRef]
- Galindo-Solís, J.M.; Fernández, F.J. Endophytic Fungal Terpenoids: Natural Role and Bioactivities. Microorganisms 2022, 10, 339. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Reported Metabolite [Reference] |
|---|---|
| Malbranchea aurantiaca | Malbrancheamide [24], Malbrancheamide B [25], Phytotoxins (1-hydroxy-2-oxoeremophil-1(10), 7(11),8(9)-trien-12(8)-olide and penicillic acid) [26], Premalbrancheamide [27] |
| Malbranchea dendritica | α-Glucosidase and Protein tyrosine phosphatase 1B (PTP-1B) [28] |
| Malbranchea filamentosa | Cytotoxic anthrasteroid glycosides, malsterosides A–C [29] |
| Malbranchea flavorosea | Polyketides (8-chloroxylarinol A, flavoroseoside) [30] |
| Malbranchea graminicola | Spiromalbramide and isomalbrancheamide B [31] |
| Malbranchea pulchella | β-glucosidase [32] |
| Malbranchea pulchella var. sulfurea | Antibiotic (Tf-26Vx) [33], β-xylosidase [34,35], Lipoamide dehydrogenase [36], Protease [37,38], Serine protease [39], Trehalase [40], Xylanase [41,42] |
| Parameter | Amount ** |
|---|---|
| Illumina reads | 8,179,242 |
| PacBio subreads | 609,002 |
| # contigs (≥0 bp) | 38 |
| Largest contig | 2,639,719 |
| Total length (≥0 bp) | 26,468,106 |
| GC (%) | 49.06 |
| N50 | 1,540,514 |
| N90 | 394,453 |
| L50 | 7 |
| L90 | 20 |
| # N’s per 100 kbp | 0.00 |
| # of rRNA | 42 |
| # of tRNA | 177 |
| Complete BUSCOs (C) * | 4653 (95.7%) |
| Complete and single-copy BUSCOs (S) * | 4615 (94.9%) |
| Complete and duplicated BUSCOs (D) * | 38 (0.8%) |
| Fragmented BUSCOs (F) * | 43 (0.9%) |
| Missing BUSCOs (M) * | 166 (3.4%) |
| Total lineage BUSCO * | 4862 |
| PFAM Domain | Endoproteases Catalyzing Keratin Hydrolysis | Exoproteases Involved in Keratin Hydrolysis | Oligopeptidases Involved in Keratin Hydrolysis | Other Enzymes Involved in Keratin Hydrolysis (Membrane Proteases) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Subtilase Family (PF00082) | Lon Protease (S16) C-terminal Proteolytic Domain (PF05362) | Insulinase (Peptidase Family M16) (PF00675) | Fungalysin Metallopeptidase (M36) (PF02128) | Prolyl Oligopeptidase Family (PF00326) | Serine Carboxypeptidase (PF00450) | Zinc Carboxypeptidase (PF00246) | Peptidase Family M28 (PF04389) | Peptidase Family M3 (PF01432) | Peptidase Family S41 (PF03572) | Metallopeptidase Family M24 (PF00557) | |||
| Onygenales | Onygenaceae | Coccidioides immitis RS | 18 | 2 | 6 | 2 | 5 | 10 | 3 | 9 | 2 | 1 | 9 |
| C. posadasii C735 | 17 | 2 | 6 | 2 | 5 | 11 | 3 | 9 | 2 | 1 | 9 | ||
| Uncinocarpus reesii UAMH 1704 | 21 | 2 | 6 | 2 | 4 | 7 | 4 | 10 | 3 | 2 | 9 | ||
| Aphanoascus verrucosus IHEM 4434 | 21 | 2 | 6 | 2 | 5 | 8 | 4 | 12 | 2 | 1 | 9 | ||
| Chrysosporium keratinophilum CBS 104.62 | 22 | 2 | 6 | 2 | 5 | 8 | 4 | 11 | 2 | 1 | 9 | ||
| Malbrancheaceae | Malbranchea zuffiana CBS 219.58 | 19 | 2 | 7 | 1 | 6 | 7 | 4 | 8 | 2 | 1 | 11 | |
| Arthrodermataceae | Trichophyton rubrum CBS 118892 | 21 | 4 | 9 | 9 | 3 | 15 | 4 | 15 | 2 | 2 | 13 | |
| T. benhamiae CBS 112371 | 17 | 2 | 7 | 5 | 4 | 11 | 5 | 11 | 2 | 2 | 8 | ||
| Nannizzia gypsea CBS 118893 | 15 | 2 | 7 | 5 | 5 | 12 | 4 | 11 | 2 | 3 | 9 | ||
| Microsporum canis CBS 113480 | 19 | 2 | 6 | 5 | 6 | 11 | 4 | 11 | 2 | 2 | 9 | ||
| Ajellomycetaceae | Paracoccidioides brasiliensis PB01 | 7 | 2 | 7 | 0 | 5 | 5 | 1 | 6 | 2 | 1 | 7 | |
| Histoplasma capsulatum G186AR | 8 | 2 | 7 | 0 | 5 | 7 | 1 | 7 | 2 | 1 | 9 | ||
| Eurotiales | Aspergillaceae | Aspergillus fumigatus Af293 | 6 | 2 | 5 | 1 | 5 | 12 | 1 | 7 | 2 | 1 | 11 |
| A. flavus NRRL3357 | 6 | 2 | 6 | 2 | 7 | 12 | 2 | 14 | 4 | 5 | 12 | ||
| A. nidulans FGSC A4 | 5 | 2 | 6 | 0 | 8 | 5 | 2 | 7 | 0 | 0 | 11 | ||
| Arachnomycetales | Arachnomycetaceae | Arachnomyces peruvianus gpAraPeru1.1 | 5 | 2 | 6 | 0 | 6 | 9 | 2 | 7 | 2 | 2 | 10 |
| PFAM domain | Cellulases | Xylanases | Pectinases | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cellulase (Glycosyl Hydrolase Family 5) (PF00150) | Glycosyl Hydrolase Family 3 N Terminal Domain (PF00933) | Glycosyl Hydrolase Family 3 C-terminal Domain (PF01915) | Glycosyl Hydrolases Family 6 (PF01341) | Glycosyl Hydrolase Family 45 (PF02015) | Glycosyl Hydrolase Family 7 (PF00840) | Glycosyl Hydrolases Family 8 (PF01270) | Glycosyl Hydrolase Family 9 (PF00759) | Glycosyl Hydrolase Family 10 (PF00331) | Glycoside Hydrolase Family 44 (PF12891) | Glycosyl Hydrolase Family 48 (PF02011) | Glycosyl Hydrolases Family 11 (PF00457) | Glycosyl Hydrolase Family 12 (PF01670) | Glycosyl Hydrolase Family 30 Beta Sandwich Domain (PF17189) | Pectate Lyase Superfamily Protein (PF12708) | Pectate Lyase (PF04431) | Pectinesterase (PF01095) | |||
| Onygenales | Onygenaceae | Coccidioides immitis RS | 1 | 5 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| C. posadasii C735 | 1 | 5 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | ||
| Uncinocarpus reesii UAMH 1704 | 1 | 5 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | ||
| Aphanoascus verrucosus IHEM 4434 | 2 | 6 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | ||
| Chrysosporium keratinophilum CBS 104.62 | 3 | 6 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | ||
| Malbrancheaceae | Malbranchea zuffiana CBS 219.58 | 2 | 7 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | |
| Arthrodermataceae | Trichophyton rubrum CBS 118892 | 2 | 5 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | |
| T. benhamiae CBS 112371 | 3 | 6 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | ||
| Nannizzia gypsea CBS 118893 | 2 | 6 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | ||
| Microsporum canis CBS 113480 | 3 | 6 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | ||
| Ajellomycetaceae | Paracoccidioides brasiliensis PB01 | 3 | 4 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 0 | 0 | |
| Histoplasma capsulatum G186AR | 3 | 4 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | ||
| Eurotiales | Aspergillaceae | Aspergillus fumigatus Af293 | 13 | 17 | 15 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 13 | 8 | 6 |
| A. flavus NRRL3357 | 14 | 24 | 22 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 6 | 13 | 6 | ||
| A. nidulans FGSC A4 | 13 | 21 | 19 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 14 | 12 | 3 | ||
| Arachnomycetales | Arachnomycetaceae | Arachnomyces peruvianus gpAraPeru1.1 | 18 | 18 | 15 | 3 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 4 | 8 | 2 |
| PFAM domain | PKS | NRPS | NRPS/PKS | Terpene | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acyl Transferase Domain (PF00698) | Beta-Ketoacyl Synthase, C-terminal Domain (PF02801) | Beta-Ketoacyl Synthase, N-terminal Domain (PF00109) | Chalcone and Stilbene Synthases, N-terminal Domain (PF00195) | Condensation Domain (PF00668) | AMP-Binding Enzyme (PF00501) | Phosphopantetheine Attachment Site (PF00550) | Terpene Synthase Family 2, C-terminal Metal Binding (PF19086) | Terpene Synthase, N-terminal Domain (PF01397) | Terpene Synthase Family, Metal Binding Domain (PF03936) | |||
| Onygenales | Onygenaceae | Coccidioides immitis RS | 13 | 13 | 13 | 1 | 23 | 48 | 34 | 1 | 0 | 0 |
| C. posadasii C735 | 11 | 12 | 12 | 1 | 23 | 49 | 33 | 1 | 0 | 0 | ||
| Uncinocarpus reesii UAMH 1704 | 9 | 9 | 8 | 1 | 29 | 57 | 31 | 0 | 0 | 0 | ||
| Aphanoascus verrucosus IHEM 4434 | 7 | 8 | 8 | 1 | 25 | 49 | 26 | 1 | 0 | 0 | ||
| Chrysosporium keratinophilum CBS 104.62 | 10 | 11 | 11 | 1 | 29 | 51 | 30 | 1 | 0 | 0 | ||
| Malbrancheaceae | Malbranchea zuffiana CBS 219.58 | 14 | 14 | 14 | 0 | 37 | 55 | 47 | 6 | 0 | 0 | |
| Arthrodermataceae | Trichophyton rubrum CBS 118892 | 13 | 16 | 17 | 1 | 48 | 78 | 51 | 3 | 0 | 0 | |
| T. benhamiae CBS 112371 | 15 | 15 | 16 | 1 | 55 | 72 | 60 | 3 | 0 | 0 | ||
| Nannizzia gypsea CBS 118893 | 21 | 22 | 23 | 1 | 47 | 69 | 51 | 3 | 0 | 0 | ||
| Microsporum canis CBS 113480 | 25 | 25 | 28 | 1 | 59 | 80 | 68 | 5 | 0 | 0 | ||
| Ajellomycetaceae | Paracoccidioides brasiliensis PB01 | 5 | 5 | 5 | 0 | 13 | 43 | 13 | 1 | 0 | 0 | |
| Histoplasma capsulatum G186AR | 5 | 5 | 6 | 0 | 20 | 49 | 22 | 0 | 0 | 0 | ||
| Eurotiales | Aspergillaceae | Aspergillus fumigatus Af293 | 16 | 17 | 18 | 0 | 54 | 74 | 59 | 1 | 0 | 0 |
| A. flavus NRRL3357 | 39 | 39 | 44 | 3 | 72 | 121 | 102 | 11 | 0 | 0 | ||
| A. nidulans FGSC A4 | 33 | 35 | 36 | 0 | 52 | 89 | 69 | 4 | 0 | 0 | ||
| Arachnomycetales | Arachnomycetaceae | Arachnomyces peruvianus gpAraPeru1.1 | 29 | 30 | 31 | 0 | 65 | 87 | 77 | 7 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granados-Casas, A.O.; Fernández-Bravo, A.; Stchigel, A.M.; Cano-Lira, J.F. Genomic Sequencing and Functional Analysis of the Ex-Type Strain of Malbranchea zuffiana. J. Fungi 2024, 10, 600. https://doi.org/10.3390/jof10090600
Granados-Casas AO, Fernández-Bravo A, Stchigel AM, Cano-Lira JF. Genomic Sequencing and Functional Analysis of the Ex-Type Strain of Malbranchea zuffiana. Journal of Fungi. 2024; 10(9):600. https://doi.org/10.3390/jof10090600
Chicago/Turabian StyleGranados-Casas, Alan Omar, Ana Fernández-Bravo, Alberto Miguel Stchigel, and José Francisco Cano-Lira. 2024. "Genomic Sequencing and Functional Analysis of the Ex-Type Strain of Malbranchea zuffiana" Journal of Fungi 10, no. 9: 600. https://doi.org/10.3390/jof10090600
APA StyleGranados-Casas, A. O., Fernández-Bravo, A., Stchigel, A. M., & Cano-Lira, J. F. (2024). Genomic Sequencing and Functional Analysis of the Ex-Type Strain of Malbranchea zuffiana. Journal of Fungi, 10(9), 600. https://doi.org/10.3390/jof10090600