
Genome Sequencing of Lentinula edodes Revealed a Genomic Variant Block Associated with a Thermo-Tolerant Trait in Fruit Body Formation
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Materials and Cultivation Conditions
2.2. Genomic DNA Extraction and Whole-Genome Sequencing
2.3. De Novo Genome Assembly and Polishing
2.4. Genome Completeness and Coverage Estimation
2.5. Genome Annotation
2.6. Discovery of Single Nucleotide Variants (SNVs)
2.7. CAPS Marker Development
3. Results
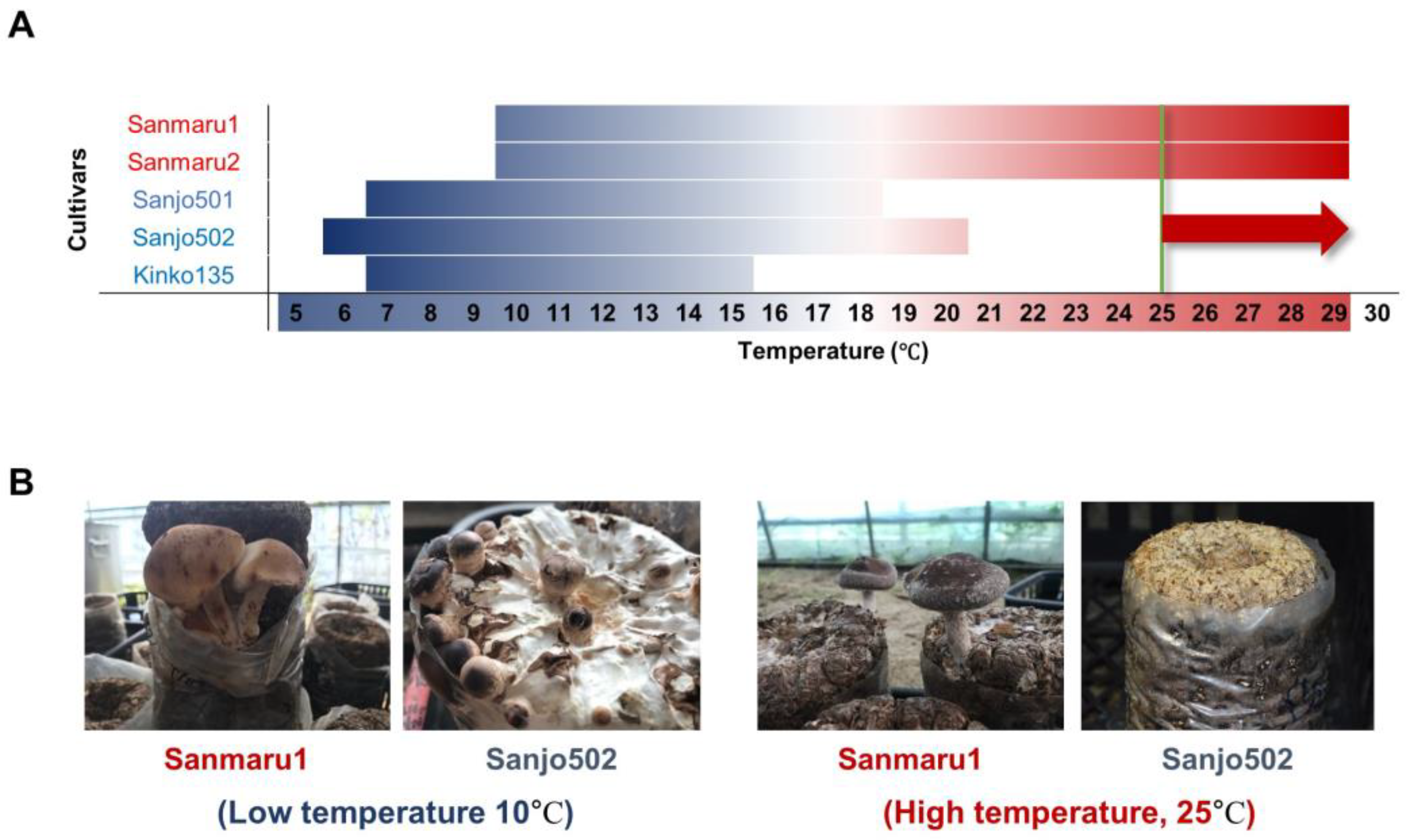
3.1. Phenotypic Characterization of High-Temperature Tolerance in Lentinula edodes Cultivars
3.2. Genetic and Genomic Basis of High-Temperature Tolerance
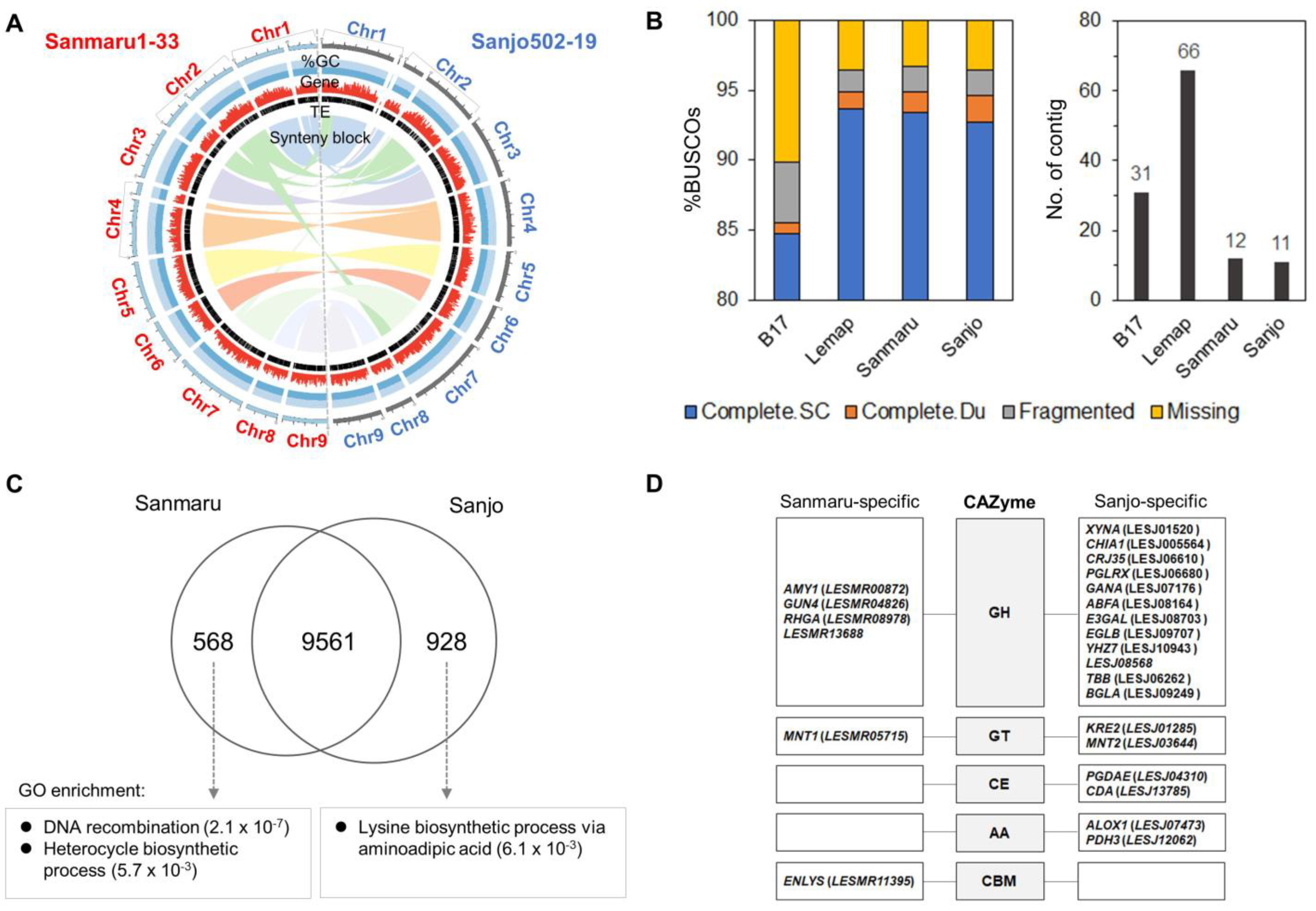
3.3. Genomic Characterization and Functional Analysis
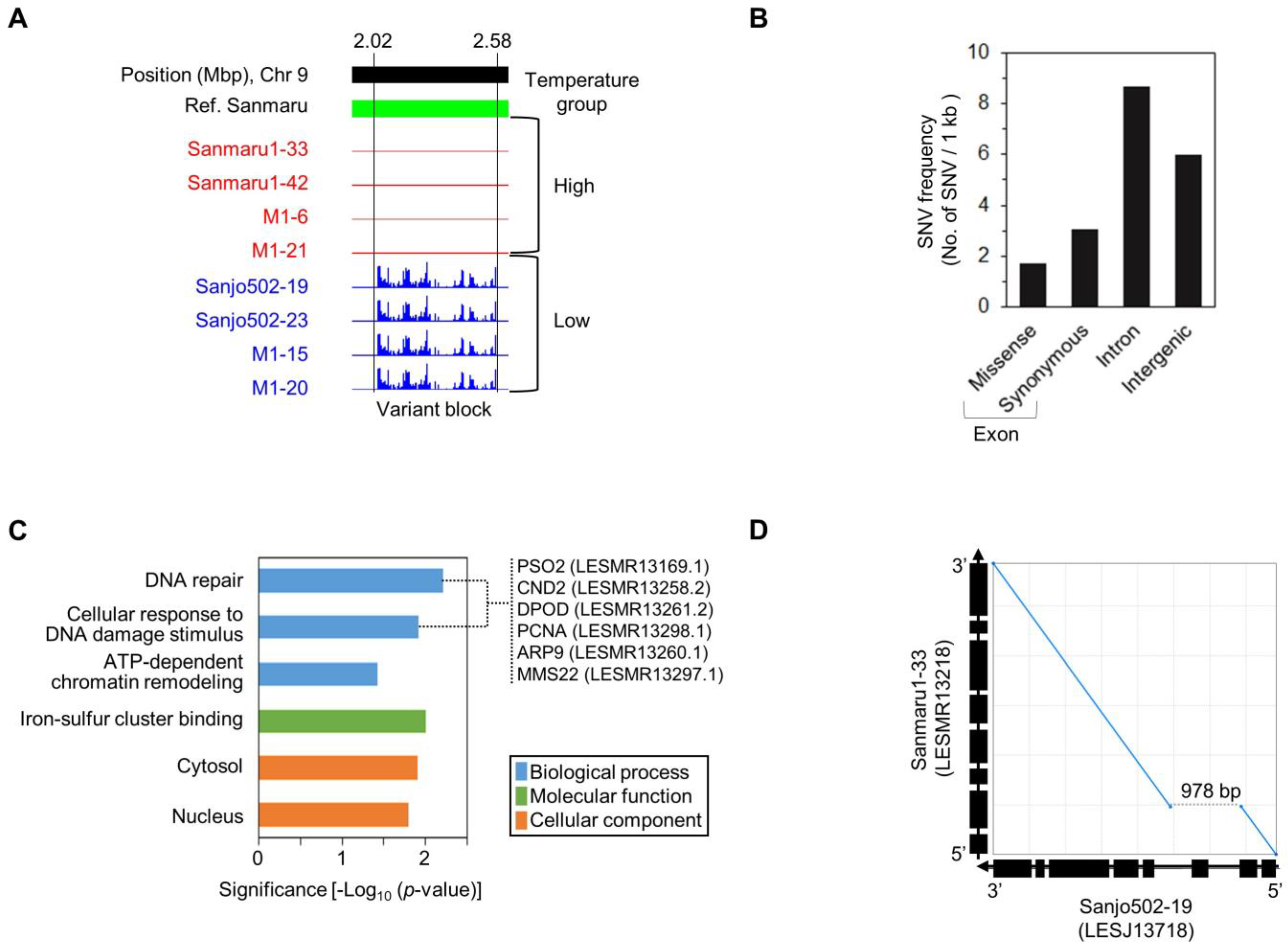
3.4. Genetic Variation and Structural Analysis Associated with High-Temperature Tolerance
3.5. Development and Validation of CAPS Markers for High-Temperature Tolerance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Royse, D.J.; Baars, J.; Tan, Q. Current Overview of Mushroom Production in the World. In Edible and Medicinal Mushrooms; John Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 5–13. [Google Scholar]
- Hasebe, K.; Ohira, I.; Arita, I. Genetic Relationship between High-, Medium- and Low-Temperature-Type Fruiting of Lentinula edodes in Wood Log Culture; Tottori Mycological Institute: Tottori, Japan, 1998. [Google Scholar]
- Leatham, G.F. Cultivation of shiitake, the Japaneseforest mushroom, on logs: A potentialindustry for the United States. For. Prod. J. 1982, 32, 29–35. [Google Scholar]
- Feder, M.E.; Hofmann, G.E. Heat-shock proteins, molecular chaperones, and the stress response: Evolutionary and ecological physiology. Annu. Rev. Physiol. 1999, 61, 243–282. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Genet. 1988, 22, 631–677. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.P.; Zhou, S.S.; Song, J.X.; Zhong, H.Y.; Zhu, T.W.; Gong, Y.H.; Zhou, Y.; Bian, Y.B. Comparative Transcriptome Analysis Provides Insights into the Mechanism by Which 2,4-Dichlorophenoxyacetic Acid Improves Thermotolerance in Lentinula edodes. Front. Microbiol. 2022, 13, 910255. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Kong, X.; Lu, Z.; Xiao, M.; Chen, M.; Zhu, L.; Shen, Y.; Hu, X.; Song, S. Para-aminobenzoic acid (PABA) synthase enhances thermotolerance of mushroom Agaricus bisporus. PLoS ONE 2014, 9, e91298. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Sato, S.; Nakade, K.; Ito, M.; Yamauchi, T.; Eda, K.; Goto, F.; Mizuno, M.; Konno, N. Screening of a Mutant That Retains Lentinan Contents Long after Being Harvested Using TILLING. ACS Agric. Sci. Technol. 2021, 1, 143–149. [Google Scholar] [CrossRef]
- Wang, G.Z.; Luo, Y.; Wang, C.; Zhou, Y.; Mou, C.Y.; Kang, H.; Xiao, Y.; Bian, Y.B.; Gong, Y.H. Hsp40 Protein LeDnaJ07 Enhances the Thermotolerance of and Regulates IAA Biosynthesis by Interacting LetrpE. Front. Microbiol. 2020, 11, 707. [Google Scholar] [CrossRef]
- Chao-Jun, M.A.; Gang-Zheng, W.; Sha-Sha, Z.; Yi, L.U.O.; Yu-Hua, G.; Yin-Bing, B. Functional analyses of anthranilate synthase gene LetrpE in Lentinula edodes by RNAi mediated gene knockdown. Mycosystema 2018, 37, 576–583. [Google Scholar] [CrossRef]
- Chen, L.; Gong, Y.; Cai, Y.; Liu, W.; Zhou, Y.; Xiao, Y.; Xu, Z.; Liu, Y.; Lei, X.; Wang, G. Genome sequence of the edible cultivated mushroom Lentinula edodes (Shiitake) reveals insights into lignocellulose degradation. PLoS ONE 2016, 11, e0160336. [Google Scholar] [CrossRef]
- Shim, D.; Park, S.-G.; Kim, K.; Bae, W.; Lee, G.W.; Ha, B.-S.; Ro, H.-S.; Kim, M.; Ryoo, R.; Rhee, S.-K. Whole genome de novo sequencing and genome annotation of the world popular cultivated edible mushroom, Lentinula edodes. J. Biotechnol. 2016, 223, 24–25. [Google Scholar] [CrossRef]
- Lefebvre, V.; Goffinet, B.; Chauvet, J.C.; Caromel, B.; Signoret, P.; Brand, R.; Palloix, A. Evaluation of genetic distances between pepper inbred lines for cultivar protection purposes: Comparison of AFLP, RAPD and phenotypic data. Theor. Appl. Genet. 2001, 102, 741–750. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef]
- Xu, X.Y.; Bai, G.H. Whole-genome resequencing: Changing the paradigms of SNP detection, molecular mapping and gene discovery. Mol. Breed. 2015, 35, 33. [Google Scholar] [CrossRef]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Song, Q.J.; Yan, L.; Quigley, C.; Jordan, B.D.; Fickus, E.; Schroeder, S.; Song, B.H.; An, Y.Q.C.; Hyten, D.; Nelson, R.; et al. Genetic Characterization of the Soybean Nested Association Mapping Population. Plant Genome-Us 2017, 10. [Google Scholar] [CrossRef]
- Brunke, S.; Hube, B. Two unlike cousins: Candida albicans and C. glabrata infection strategies. Cell. Microbiol. 2013, 15, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Zhang, L.; Gong, W.; Li, C.; Shen, N.; Gui, Y.; Bian, Y.; Kwan, H.S.; Cheung, M.K.; Xiao, Y. RNA-Seq-based high-resolution linkage map reveals the genetic architecture of fruiting body development in shiitake mushroom, Lentinula edodes. Comput. Struct. Biotechnol. J. 2021, 19, 1641–1653. [Google Scholar] [CrossRef] [PubMed]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Bruna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 2021, 3, lqaa108. [Google Scholar] [CrossRef]
- Park, S.G.; Yoo, S.I.; Ryu, D.S.; Lee, H.; Ahn, Y.J.; Ryu, H.; Ko, J.; Hong, C.P. Long-read transcriptome data for improved gene prediction in Lentinula edodes. Data Brief 2017, 15, 454–458. [Google Scholar] [CrossRef]
- Gremme, G.; Brendel, V.; Sparks, M.E.; Kurtz, S. Engineering a software tool for gene structure prediction in higher organisms. Inf. Softw. Technol. 2005, 47, 965–978. [Google Scholar] [CrossRef]
- Keller, O.; Kollmar, M.; Stanke, M.; Waack, S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 2011, 27, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Mulder, N.; Apweiler, R. InterPro and InterProScan: Tools for protein sequence classification and comparison. Methods Mol. Biol. 2007, 396, 59–70. [Google Scholar] [CrossRef]
- Drula, E.; Garron, M.L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed]
- Soderlund, C.; Nelson, W.; Shoemaker, A.; Paterson, A. SyMAP: A system for discovering and viewing syntenic regions of FPC maps. Genome Res. 2006, 16, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Salzberg, S.L.; Phillippy, A.M. Using MUMmer to identify similar regions in large sequence sets. In Current Protocols in Bioinformatics; John Wiley & Sons: Hoboken, NJ, USA, 2003; Chapter 10. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef]
- Tang, H.; Krishnakumar, V.; Zeng, X.; Xu, Z.; Taranto, A.; Lomas, J.S.; Zhang, Y.; Huang, Y.; Wang, Y.; Yim, W.C.; et al. JCVI: A versatile toolkit for comparative genomics analysis. iMeta 2024, 3, e211. [Google Scholar] [CrossRef]
- Branzei, D.; Szakal, B. DNA damage tolerance by recombination: Molecular pathways and DNA structures. DNA Repair. 2016, 44, 68–75. [Google Scholar] [CrossRef]
- Fazius, F.; Shelest, E.; Gebhardt, P.; Brock, M. The fungal alpha-aminoadipate pathway for lysine biosynthesis requires two enzymes of the aconitase family for the isomerization of homocitrate to homoisocitrate. Mol. Microbiol. 2012, 86, 1508–1530. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Li, D.D.; Wang, J.L.; Liu, Y.; Li, Y.Z.; Zhang, Z. Expanded analyses of the functional correlations within structural classifications of glycoside hydrolases. Comput. Struct. Biotechnol. J. 2021, 19, 5931–5942. [Google Scholar] [CrossRef]
- Stamets, P. Growing Gourmet and Medicinal Mushrooms = [Shokuyō oyobi yakuyō kinoko no saibai], 3rd ed.; Ten Speed Press: Berkeley, CA, USA, 2000; p. xviii. 574p. [Google Scholar]
- Sul, J.H.; Martin, L.S.; Eskin, E. Population structure in genetic studies: Confounding factors and mixed models. PLoS Genet. 2018, 14, e1007309. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.R.; Lübberstedt, T. Functional markers in plants. Trends Plant Sci. 2003, 8, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Hayward, A.C.; Tollenaere, R.; Dalton-Morgan, J.; Batley, J. Molecular marker applications in plants. Methods Mol. Biol. 2015, 1245, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Chen, C.R.; Chang, E.C. Fission yeast Ras1 effector Scd1 interacts with the spindle and affects its proper formation. Genetics 2000, 156, 995–1004. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quantification | ||
|---|---|---|
| Sanmaru 1-33 | Sanjo 502-19 | |
| Total no. of gene models predicted | 15,900 | 16,380 |
| Unique gene models (No.) | 13,726 | 14,247 |
| Genes with isoforms (No.) | 2174 | 2133 |
| Average gene length (bp) | 2059 bp | 1942 bp |
| Total bases of gene models (Mbp) | 32.74 Mbp | 31.81 Mbp |
| %Genes in the draft genome | 69.49 | 68.41 |
| No. of exon | 88,779 | 91,869 |
| Average no. of exon per gene | 5.58 | 5.60 |
| Average exon length (bp) | 285 bp | 274 bp |
| %Exon fraction in the draft genome | 53.75 | 54.28 |
| No. of intron | 72,879 | 75,489 |
| Average no. of intron per gene | 4.58 | 4.60 |
| Average intron length (bp) | 101 bp | 86 bp |
| %Intron fraction in the draft genome | 15.74 | 14.13 |
| Functional annotated genes | 13,331 (97.12%) | 13,851 (97.22%) |
| No. hit to UniProt | 7554 (55.03%) | 7718 (54.11%) |
| No. hit to NCBI NR | 13,032 (94.94%) | 13,541 (95.04%) |
| No. hit to InterPro | 12,120 (88.29%) | 12,487 (87.64%) |
| Primer Name | Sequence (5′-3′) | Product Size (bp) | Restriction Enzyme | Digested Fragment Size (bp) | |
|---|---|---|---|---|---|
| High-Temperature Sensitive | High-Temperature Tolerant | ||||
| RL-LE-306 F | GGTAACAGTCGTCAGGTGAAAG | 234 | HpaII (+) | 234 | 89/145 |
| RL-LE-306 R | CTCCGGGATCAAAGGTTACTAC | ||||
| RL-LE-307 F | GAACCTAGCTAGACTGACGCAT | 304 | ScrFI (−) | 74/230 | 304 |
| RL-LE-307 R | GATAGATAGTCATACGGCCCTC | ||||
| RL-LE-310 F | ATTCGACTCTCTCTCTTCCTCC | 326 | AluI (+) | 326 | 112/214 |
| RL-LE-310 R | CTTCTTGTCCACTCTGAGTTCC | ||||
| RL-LE-311 F | GACTAGCAACAGGTTACCTCTCC | 330 | AluI (−) | 141/189 | 330 |
| RL-LE-311 R | CCTATACCACGAGAGAAGAGTAGG | ||||
| RL-LE-312 F | GACAGCCTACATGGTGATGAG | 329 | BglII (−) | 132/197 | 329 |
| RL-LE-312 R | CTAGTGTCAAGAATGCACTCCC | ||||
| RL-LE-314 F | GGGATACTACTTCTCTCCAACG | 395 | BglII (+) | 395 | 179/216 |
| RL-LE-314 R | CTACCGGACGACTGAACTTCT | ||||
| RL-LE-316 F | CTACACGCCTAGCTCAGAAGTT | 192 | NlaIII (−) | 36/61/95 | 36/156 |
| RL-LE-316 R | CTAACAGCTCTAGGATAGGCAGAC | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, S.-i.; Moon, S.; Hong, C.P.; Park, S.-G.; Shim, D.; Ryu, H. Genome Sequencing of Lentinula edodes Revealed a Genomic Variant Block Associated with a Thermo-Tolerant Trait in Fruit Body Formation. J. Fungi 2024, 10, 628. https://doi.org/10.3390/jof10090628
Yoo S-i, Moon S, Hong CP, Park S-G, Shim D, Ryu H. Genome Sequencing of Lentinula edodes Revealed a Genomic Variant Block Associated with a Thermo-Tolerant Trait in Fruit Body Formation. Journal of Fungi. 2024; 10(9):628. https://doi.org/10.3390/jof10090628
Chicago/Turabian StyleYoo, Seung-il, Suyun Moon, Chang Pyo Hong, Sin-Gi Park, Donghwan Shim, and Hojin Ryu. 2024. "Genome Sequencing of Lentinula edodes Revealed a Genomic Variant Block Associated with a Thermo-Tolerant Trait in Fruit Body Formation" Journal of Fungi 10, no. 9: 628. https://doi.org/10.3390/jof10090628