
A Genomic Sequence Resource of Diaporthe mahothocarpus GZU-Y2 Causing Leaf Spot Blight in Camellia oleifera


Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Material and Culture Conditions
2.2. DNA Extraction
2.3. Genome Sequencing and Assembly
2.4. Phylogenetic Analysis
2.5. Genome Prediction
2.6. Gene Function Annotation
2.7. Data Availability
3. Results
3.1. Genome Assembly and Genomic Characteristics
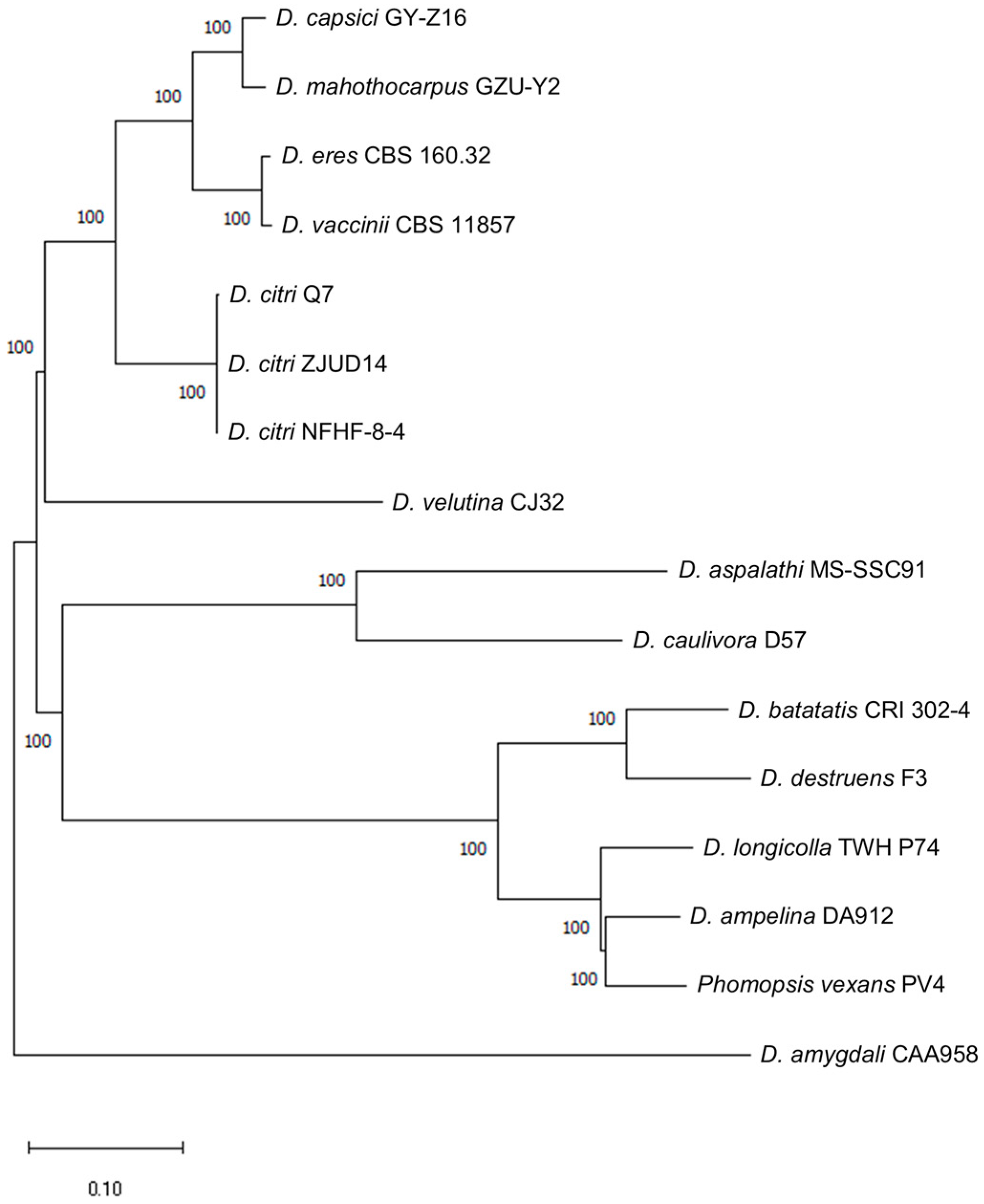
3.2. Phylogenetic Analysis
3.3. Gene Prediction
3.3.1. Prediction of Protein-Coding Genes
3.3.2. Prediction of Non-Coding RNA
3.3.3. Prediction of Pseudogenes
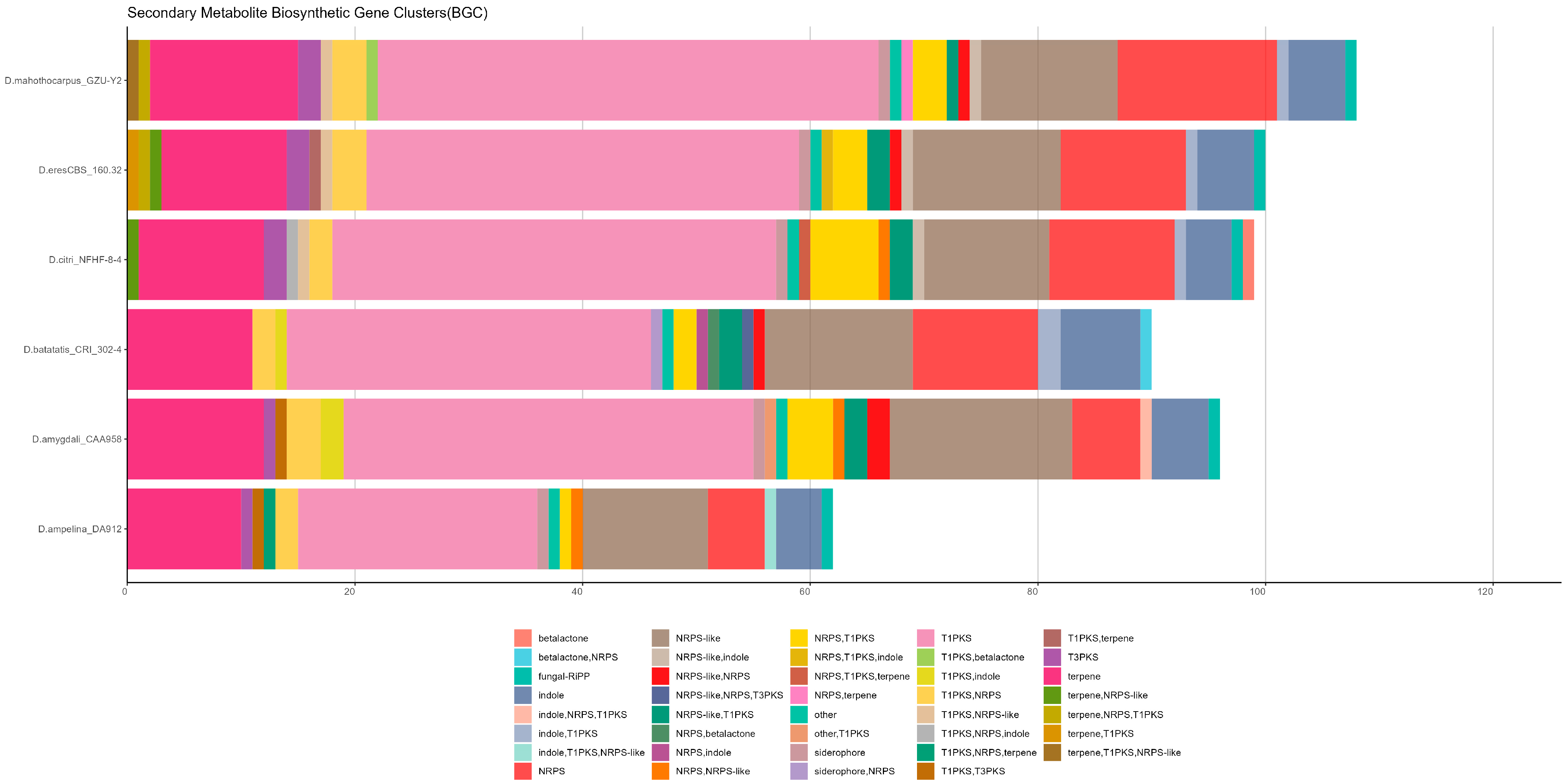
3.3.4. Prediction of Gene Cluster
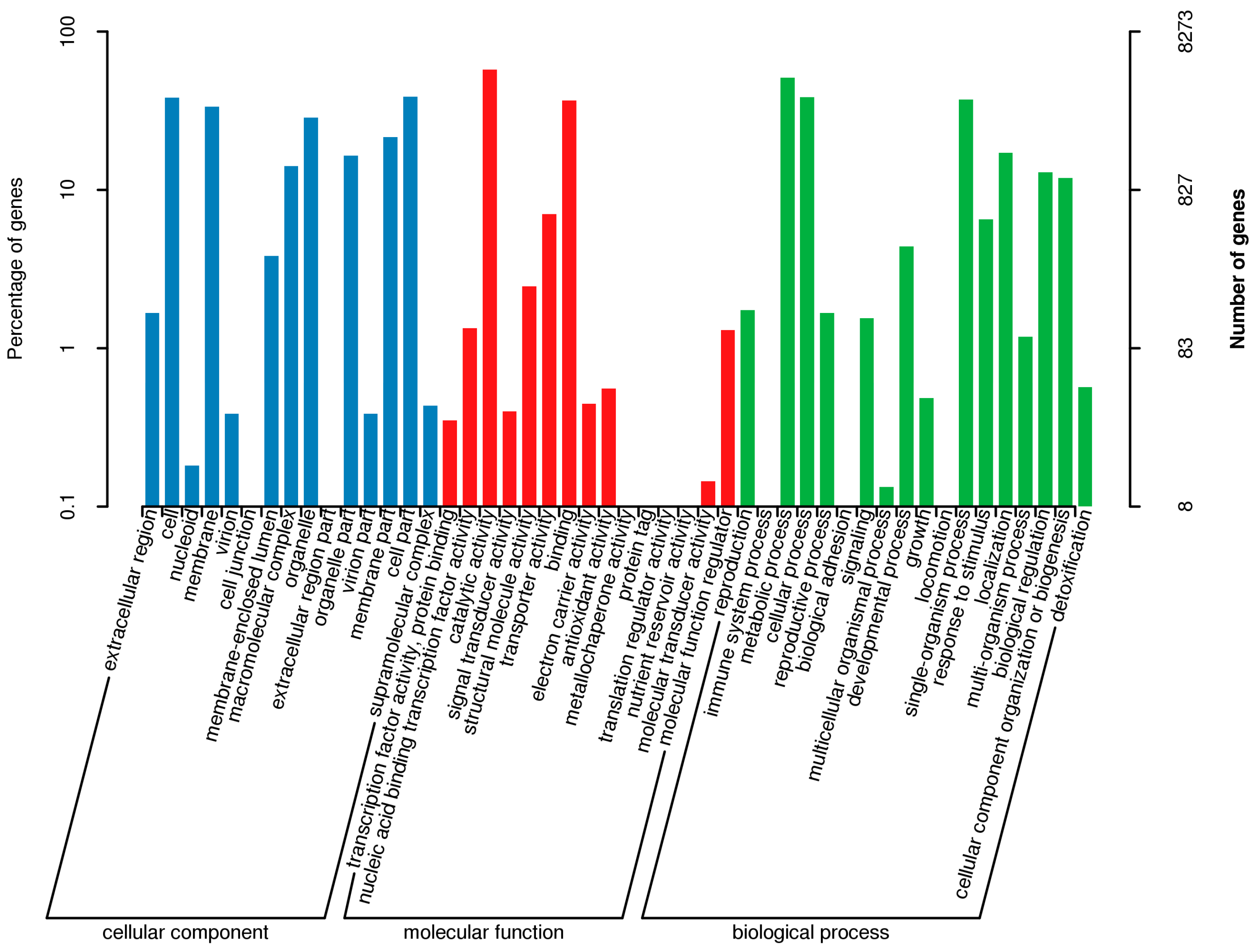
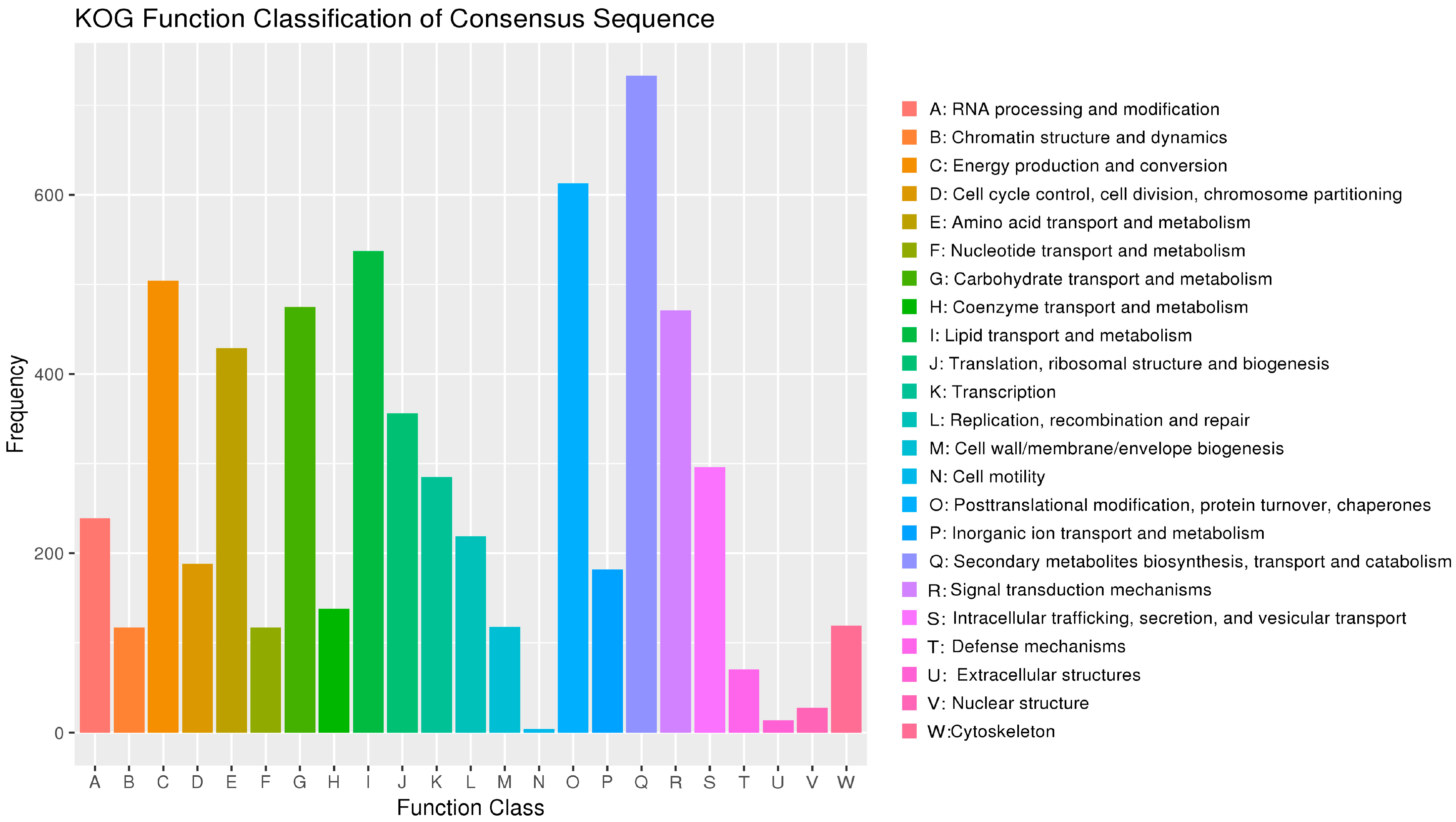
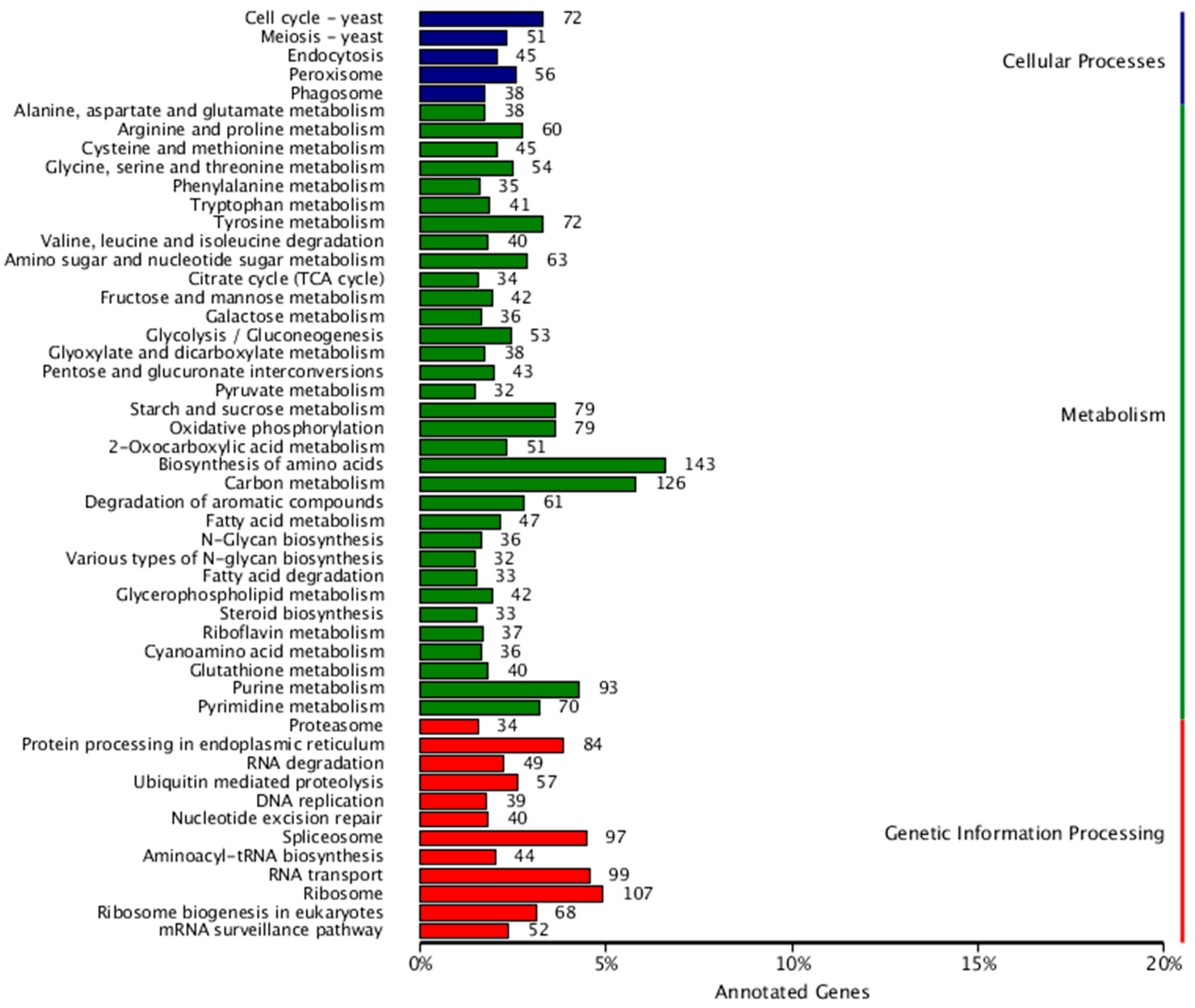
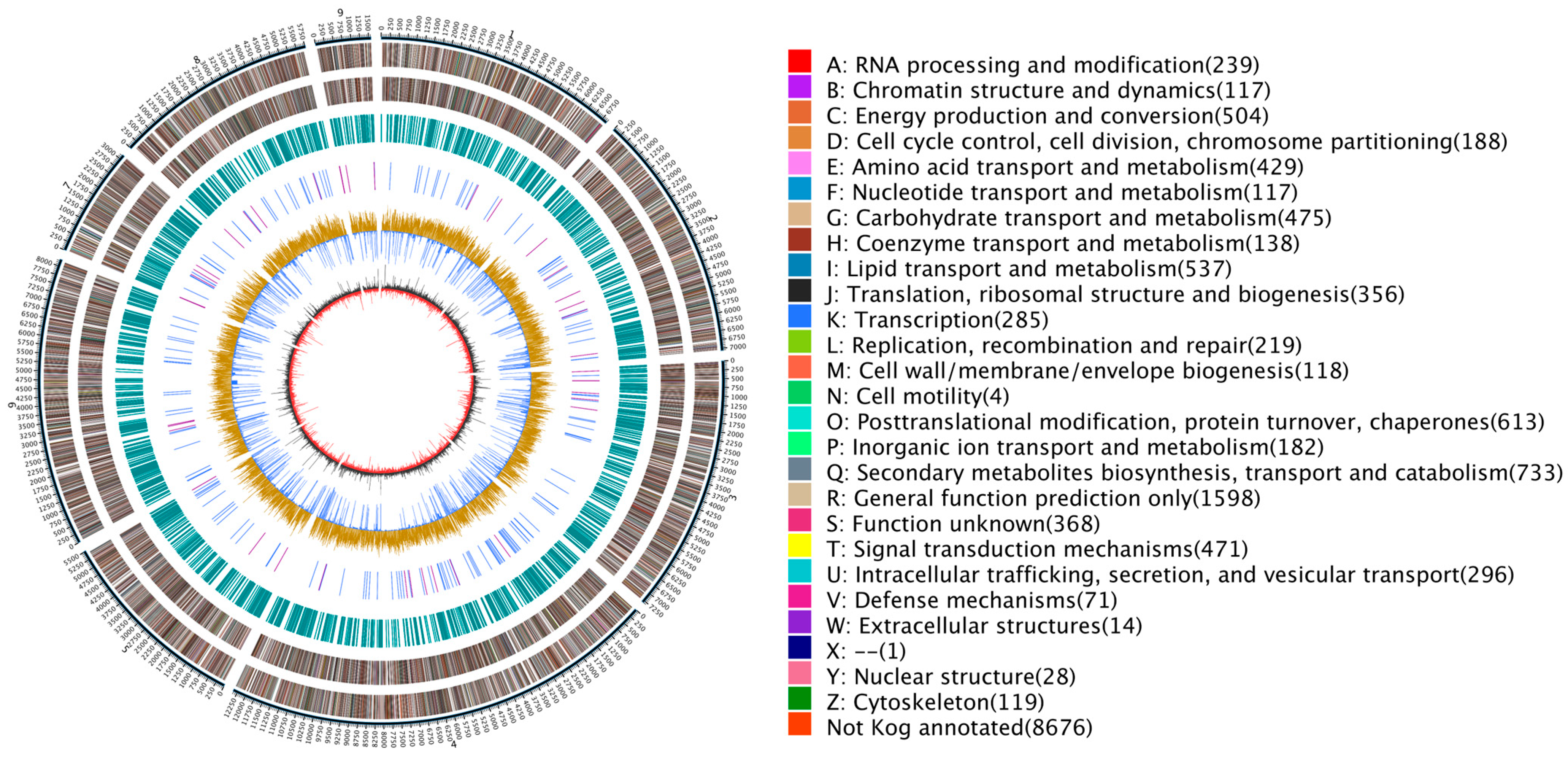
3.4. Gene Annotation
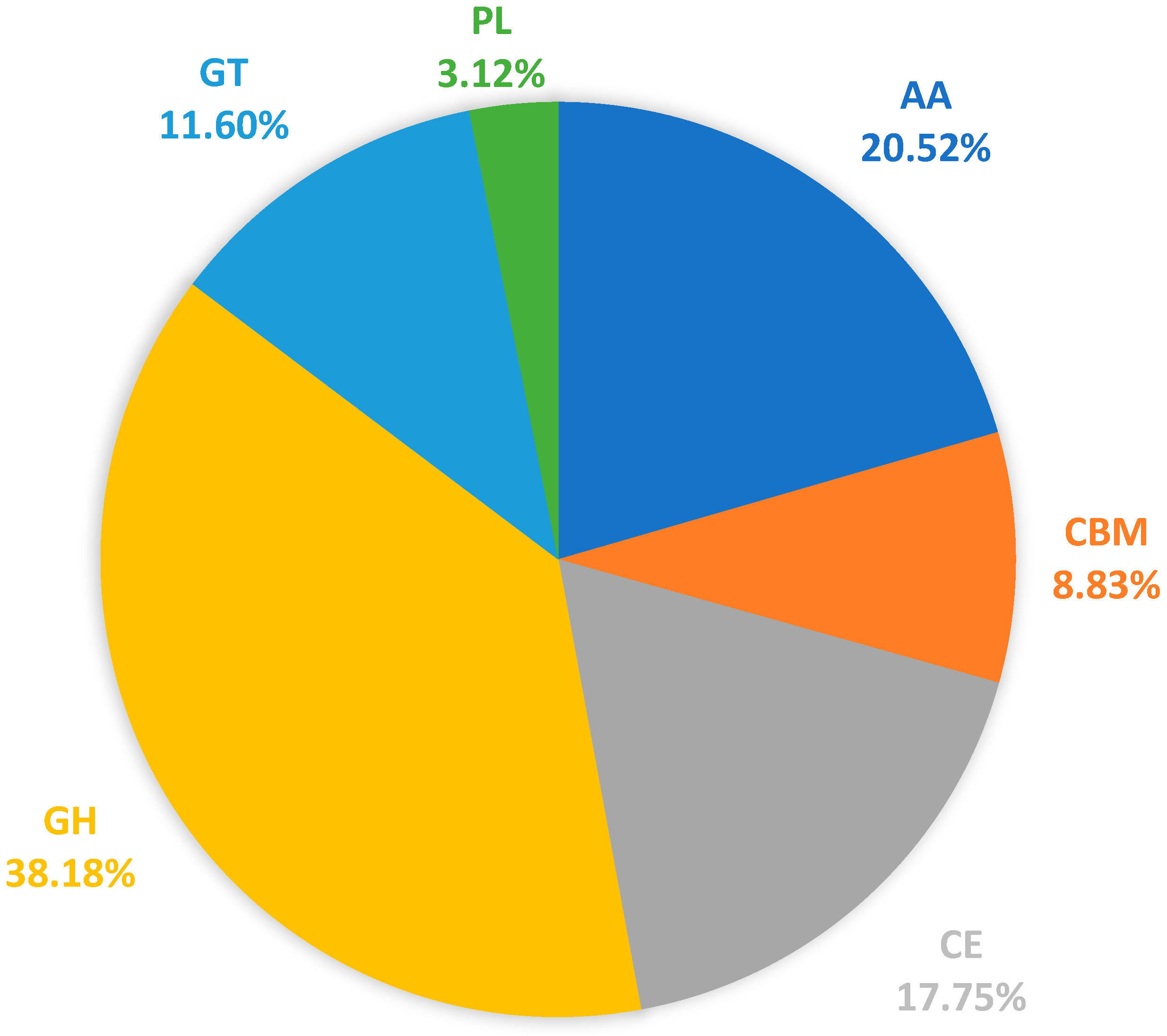
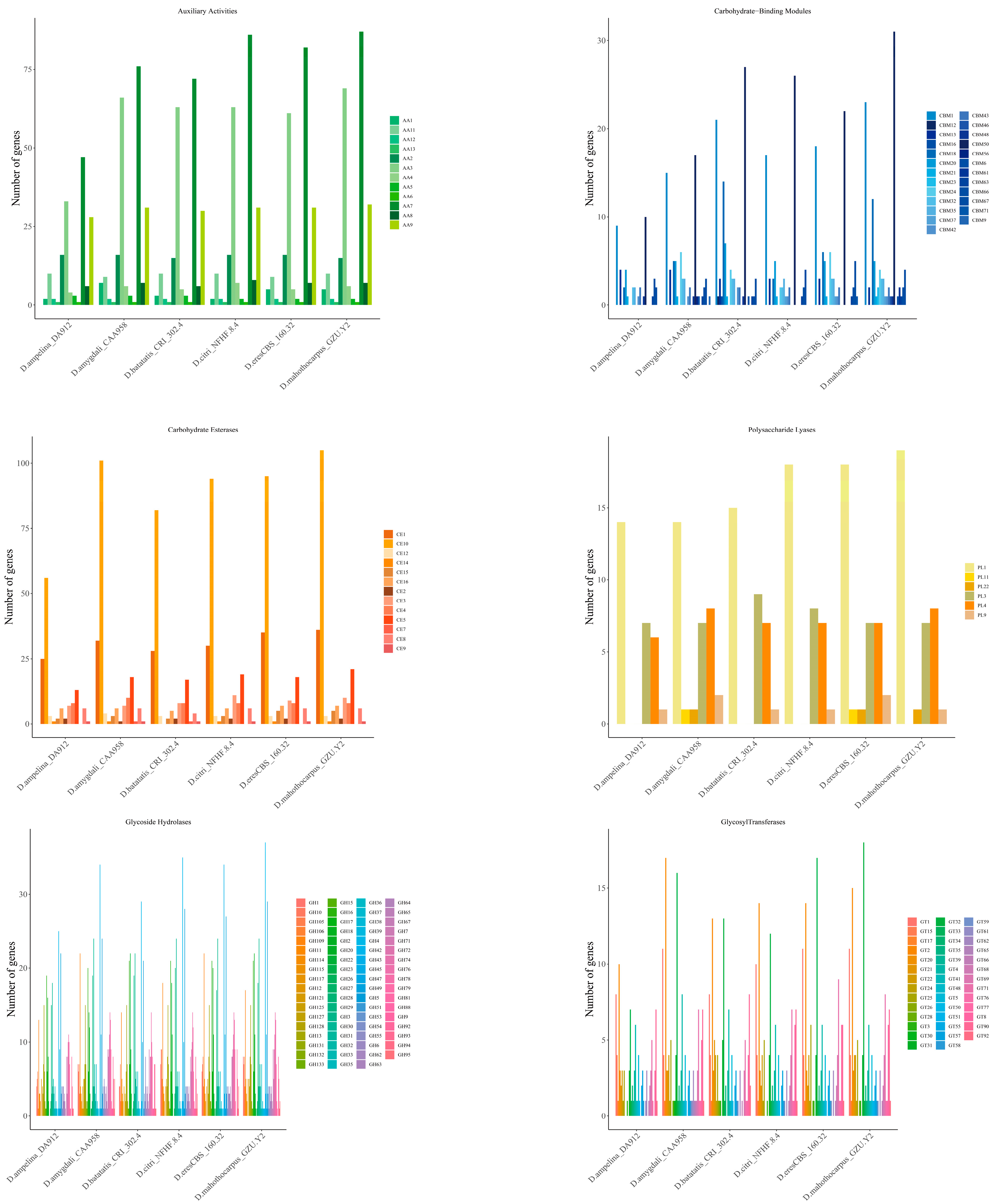
3.5. Carbohydrate-Active Enzymes (CAZymes)
3.6. Pathogenic System Analysis
3.7. Analysis of Protein Subcellular Localization
3.8. Comparative Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dissanayake, A.J.; Liu, M.; Zhang, W.; Chen, Z.; Udayanga, D.; Chukeatirote, E.; Li, X.; Yan, J.; Hyde, K.D. Morphological and molecular characterisation of Diaporthe species associated with grapevine trunk disease in China. Fungal Biol. 2015, 119, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.H. Research on plant source agents for major diseases of oil tea. Zhongnan Univ. For. Sci. Technol. 2011, 19, 78–87. [Google Scholar]
- Xiao, Y.; Huo, G.; Liu, L.; Yang, C.; Cui, C. First report of postharvest fruit rot disease of yellow peach caused by Diaporthe eres in China. Plant Dis. 2022, 106, 1983. [Google Scholar] [CrossRef]
- Yang, Q.; Tang, J.; Zhou, G.Y. Characterization of Diaporthe species on Camellia oleifera in Hunan Province, with descriptions of two new species. MycoKeys 2021, 84, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhang, Y.; Wang, X.; Pan, Y.; Yu, C.; Yang, J. First Report of Leaf Spot Blight of Camellia oleifera Caused by Diaporthe mahothocarpus in China. Plant Dis. 2024, 108, 516. [Google Scholar] [CrossRef]
- Udayanga, D.; Liu, X.; Crous, P.W.; McKenzie, E.H.; Chukeatirote, E.; Hyde, K.D. A muli-locus phylogenetic evaluation of Diaporthe (Phomopsi). Fungal Divers. 2012, 56, 157–171. [Google Scholar] [CrossRef]
- Santos, J.M.; Correia, V.G.; Phillips, A.J. Primers for mating type dagnosis in Diaporthe and Phomopsis: Their use in teleomorph induction in vitro and biological species definition. Fungal Biol. 2010, 114, 255–270. [Google Scholar] [CrossRef]
- Erincik, O.; Madden, L.V. Effect of growth stage on susceptibility of grape berry and rachis tissues to infection by Phomopsis viticola. Plant Dis. 2001, 85, 517–520. [Google Scholar] [CrossRef]
- Guarnaccia, V.; Crous, P.W. Emerging citrus diseases in Europe caused by species of Diaporthe. IMA Fungus 2017, 8, 317–334. [Google Scholar] [CrossRef]
- Mondal, S.N. Saprophytic colonization of citrus twigs by Diaporthe citri and factors affecting pycnidial production and conidial survivalc. Plant Dis. 2007, 91, 387–392. [Google Scholar] [CrossRef]
- Thompson, S.; Tan, Y.; Young, A.; Neate, S.; Aitken, E.; Shivas, R. Stem cankers on sunflower (Helianthus annuus) in Australia reveal a complex of pathogenic Diaporthe (Phomopsis) species. Persoonia 2011, 27, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Guarnaccia, V.; Groenewald, J.Z.; Woodhall, J.; Armengol, J.; Cinelli, T.; Eichmeier, A.; Ezra, D.; Fontaine, F.; Gramaje, D.; Gutierrez-Aguirregabiria, A.; et al. Diaporthe diversity and pathogenicity revealed from a broad survey of grapevine diseases in Europe. Persoonia 2018, 40, 135–153. [Google Scholar] [CrossRef]
- Udayanga, D.; Castlebury, L.A.; Rossman, A.Y.; Chukeatirote, E.; Hyde, K.D. The Diaporthe sojae species complex: Phylogenetic re-assessment of pathogens associated with soybean, cueurbits and other field crops. Fungal Biol. 2015, 119, 383–407. [Google Scholar] [CrossRef]
- Fu, F.-F.; Hao, Z.; Wang, P.; Lu, Y.; Xue, L.-J.; Wei, G.; Tian, Y.; Hu, B.; Xu, H.; Shi, J.; et al. Genome Sequence and Comparative Analysis of Colletotrichum gloeosporioides Isolated from Liriodendron Leaves. Phytopathology 2020, 110, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef]
- Han, Y.; Wessler, S.R. MITE-Hunter: A program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 2010, 38, e199. [Google Scholar] [CrossRef]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, i351–i358. [Google Scholar] [CrossRef]
- Edgar, R.C.; Myers, E.W. PILER: Identification and classification of genomic repeats. Bioinformatics 2005, 21, i152–i158. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Chen, N. Using Repeat Masker to Identify Repetitive Elements in Genomic Sequences. In Current Protocols in Bioinformatics; Baxevanis, A.D., Ed.; Wiley: Hoboken, NJ, USA, 2004; Chapter 4: Unit 4.10. [Google Scholar]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19, ii225. [Google Scholar] [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Blanco, E.; Parra, G.; Guigó, R. Using geneid to identify genes. Curr. Protoc. Bioinform. 2007, 18, 4.3.1–4.3.28. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef]
- Keilwagen, J.; Wenk, M.; Erickson, J.L.; Schattat, M.H.; Grau, J.; Hartung, F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016, 44, e89. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.A.; Haas, B.J.; Hamilton, J.P.; Mount, S.M.; Buell, C.R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genom. 2006, 7, 327. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; Van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Shi, X.L.; Yang, J.; Zhang, Y.; Qin, P.; Zhou, H.Y.; Chen, Y.Z. The photoactivated antifungal activity and possible mode of action of sodium pheophorbide a on Diaporthe mahothocarpus causing leaf spot blight in Camellia oleifera. Front. Microbiol. 2024, 15, 1403478. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Gardiner, D.M.; Dodds, P.N.; Tini, F.; Covarelli, L.; Singh, K.B.; Manners, J.M.; Taylor, J.M. EffectorP: Predicting Fungal Effector Proteins from Secretomes Using Machine Learning. New Phytol. 2015, 210, 743–761. [Google Scholar] [CrossRef]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32 (Suppl. 1), D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Janan, T.E.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Saier, M.H., Jr.; Tran, C.V.; Barabote, R.D. TCDB: The Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 2006, 34 (Suppl. 1), D181–D186. [Google Scholar] [CrossRef]
- Winnenburg, R.; Baldwin, T.K.; Urban, M.; Rawlings, C.; Köhler, J.; Hammond-Kosack, K.E. PHI-base: A new database for pathogen host interactions. Nucleic Acids Res. 2006, 34 (Suppl. 1), D459–D464. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37 (Suppl. 1), D233–D238. [Google Scholar] [CrossRef]
- Rafiei, V.; Velez, H.; Tzelepis, G. The Role of Glycoside Hydrolases in Phytopathogenic Fungi and Oomycetes Virulence. Int. J. Mol. Sci. 2021, 22, 9359. [Google Scholar] [CrossRef]
- Kubicek, C.P.; Starr, T.L.; Glass, N.L. Plant cell wall-degrading enzymes and their secretion in plant-pathogenic fungi. Annu. Rev. Phytopathol. 2014, 52, 427–451. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; De Silva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. PHI-base: The pathogen-host interactions database. Nucleic Acids Res. 2019, 48, D613–D620. [Google Scholar] [CrossRef]
- Liu, J.; Wei, Y.; Yin, Y.; Zhu, K.; Liu, Y.; Ding, H.; Lei, J.; Zhu, W.; Zhou, Y. Effects of Mixed Decomposition of Pinus sylvestris var. mongolica and Morus alba Litter on Microbial Diversity. Microorganisms 2022, 10, 1117. [Google Scholar] [PubMed]
- Chandrasekaran, M.; Thangavelu, B.; Chun, S.C.; Sathiyabama, M. Proteases from phytopathogenic fungi and their importance in phytopathogenicity. J. Gen. Plant Pathol. 2016, 82, 233–239. [Google Scholar] [CrossRef]
- Garcia, J.F.; Lawrence, D.P.; Morales-Cruz, A.; Travadon, R.; Minio, A.; Hernandez-Martinez, R.; Rolshausen, P.E.; Baumgartner, K.; Cantu, D. Phylogenomics of Plant-Associated Botryosphaeriaceae Species. Front. Microbiol. 2021, 12, 652802. [Google Scholar] [CrossRef] [PubMed]
- Hilário, S.; Gonçalves, M.F.; Fidalgo, C.; Tacão, M.; Alves, A. Genome Analyses of Two Blueberry Pathogens: Diaporthe amygdali CAA958 and n CBS 160.32. J. Fungi 2022, 8, 804. [Google Scholar] [CrossRef]
- Li, S.; Song, Q.; Martins, A.M.; Cregan, P. Draft genome sequence of Diaporthe aspalathi isolate MS-SSC91, a fungus causing stem canker in soybean. Genom. Data 2016, 7, 262–263. [Google Scholar] [CrossRef]
- Gai, Y.; Xiong, T.; Xiao, X.; Li, P.; Zeng, Y.; Li, L.; Riely, B.K.; Li, H. The Genome Sequence of the Citrus Melanose Pathogen Diaporthe citri and Two Citrus related Diaporthe species. Phytopathology 2020, 111, 779–783. [Google Scholar] [CrossRef]
- Liu, X.Y.; Chaisiri, C.; Lin, Y.; Yin, W.X.; Luo, C.X. Whole-genome sequence of Diaporthe citri isolate NFHF-8-4, the causal agent of citrus melanose. Mol. Plant Microbe Interact. 2021, 34, 845–847. [Google Scholar] [CrossRef]
- Fang, X.; Qin, K.; Li, S.; Han, S.; Zhu, T. Whole genome sequence of Diaporthe capsici, a new pathogen of walnut blight. Genomics 2020, 112, 3751–3761. [Google Scholar] [CrossRef]
- Baroncelli, R.; Amby, D.B.; Zapparata, A.; Sarrocco, S.; Vannacci, G.; Le Floch, G.; Harrison, R.J.; Holub, E.; Sukno, S.A.; Sreenivasaprasad, S.; et al. Gene family expansions and contractions are associated with host range in plant pathogens of the genus Colletotrichum. BMC Genom. 2016, 17, 555. [Google Scholar] [CrossRef]
- Kong, L.; Chen, J.; Dong, K.; Shafik, K.; Xu, W. Genomic analysis of Colletotrichum camelliae responsible for tea brown blight disease. BMC Genom. 2023, 24, 528. [Google Scholar] [CrossRef]
- Mena, E.; Garaycochea, S.; Stewart, S.; Montesano, M.; Ponce De León, I. Comparative genomics of plant pathogenic Diaporthe species and transcriptomics of Diaporthe caulivora during host infection reveal insights into pathogenic strategies of the genus. BMC Genom. 2022, 23, 175. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.-M.; Huang, Y.-Y.; Liang, M.-H. Analysis of the probiotic characteristics and adaptability of Lactiplantibacillus plantarum DMDL 9010 to gastrointestinal environment by complete genome sequencing and corresponding phenotypes. LWT 2022, 158, 113129. [Google Scholar] [CrossRef]
- Bradley, E.L.; Ökmen, B.; Doehlemann, G.; Henrissat, B.; Bradshaw, R.E.; Mesarich, C.H. Secreted Glycoside Hydrolase Proteins as Effectors and Invasion Patterns of Plant-Associated Fungi and Oomycetes. Front. Plant Sci. 2022, 13, 853106. [Google Scholar] [CrossRef] [PubMed]
- Črešnar, B.; Petrič, Š. Cytochrome P450 enzymes in the fungal kingdom. Biochim. Biophys. Acta 2011, 1814, 29–35. [Google Scholar] [CrossRef]
- Mair, W.J.; Deng, W.; Mullins, J.G.; West, S.; Wang, P.; Besharat, N.; Ellwood, S.R.; Oliver, R.P.; Oliver, R.P.; Lopez-Ruiz, F.J.; et al. Demethylase inhibitor fungicide resistance in Pyrenophora teres f. sp. Teres associated with target site modification and inducible overexpression of CYP51. Front. Microbiol. 2016, 7, 1279. [Google Scholar]
- Agrawal, Y.; Khatri, I.; Subramanian, S.; Shenoy, B.D. Genome Sequence, Comparative Analysis, and Evolutionary Insights into Chitinases of Entomopathogenic Fungus Hirsutella thompsonii. Genome Biol. Evol. 2015, 7, 916–930. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Values | |
|---|---|---|
| Reads features (PacBio) | Total read number (G) | 6.92040043 |
| SeqNum | 809,585 | |
| SumBase (G) | 6.92040043 | |
| N50Len | 9545 | |
| MeanLen | 8548 | |
| MaxLen | 45,409 | |
| Genome features | Predicted genome size (Mbp) | 58.97 |
| Complete BUSCOs (%) | 97.93 | |
| Complete and single-copy BUSCOs (%) | 97.24 | |
| Complete and duplicated BUSCOs (%) | 0.69 | |
| Fragmented BUSCOs (%) | 0 | |
| Missing BUSCOs (%) | 2.07 | |
| Total Lineage BUSCOs | 290 | |
| GC content (%) | 50.65 | |
| Contig Length (bp) | 58,973,678 | |
| Contig Number | 62 | |
| Contig N50 (bp) | 7,066,871 | |
| Contig N90 (bp) | 5,516,847 | |
| Gaps Number | 0 | |
| Repeat sequence (%) | 3.22 | |
| Protein-coding genes | 15,918 | |
| Number of non-coding RNA | 577 | |
| Pseudogene number | 4 | |
| Protein Sequence and Transporter Protein Classification Database (TCDB) | 135 | |
| Pathogen host interactive genes | 4879 | |
| Cytochrome p450 Engineering Database | 909 | |
| Fungal virulence factors | 3371 | |
| Carbohydrate-active enzymes | 1058 | |
| Signal peptide | 1919 | |
| Transmembrane protein | 3467 | |
| Secreted protein | 1431 | |
| Effector protein | 164 |
| Type | Number | Length (bp) | Percentage (%) |
|---|---|---|---|
| ClassI | 246 | 593,229 | 1.01 |
| ClassI/LINE | 20 | 1337 | 0.00 |
| ClassI/LTR/Copia | 70 | 106,154 | 0.18 |
| ClassI/LTR/Gypsy | 28 | 3848 | 0.01 |
| ClassI/PLE|LARD | 57 | 209,844 | 0.36 |
| ClassI/TRIM | 11 | 3783 | 0.01 |
| ClassI/Unknown | 60 | 268,324 | 0.45 |
| ClassII | 153 | 16,242 | 0.03 |
| ClassII/Helitron | 5 | 381 | 0.00 |
| ClassII/MITE | 45 | 9075 | 0.02 |
| ClassII/TIR | 85 | 5974 | 0.01 |
| ClassII/Unknown | 18 | 1183 | 0.00 |
| PotentialHostGene | 58 | 480,745 | 0.82 |
| SSR | 738 | 354,092 | 0.60 |
| Unknown | 1693 | 571,362 | 0.97 |
| Total | 1195 | 1,901,760 | 3.22 |
| Features | Values |
|---|---|
| Number of protein-coding genes | 15,918 |
| Total length of protein-coding genes | 33,450,242 |
| Average length of protein-coding genes | 2101.41 |
| Total exon length | 29,284,507 |
| Average length of exons | 623.78 |
| Number of exons | 46,947 |
| Average number of exons per gene | 2.95 |
| Total length of CDS | 23,415,588 |
| Average length of CDS | 506.72 |
| Number of CDS | 46,210 |
| Average number of CDSs per gene | 2.9 |
| Total length of intron | 4,165,735 |
| Average length of intron | 134.25 |
| Number of introns | 31,029 |
| Average number of introns per gene | 1.95 |
| Scaffold ID | Gene Cluster | Start | End | Length (bp) | Type | Most Similar Known Cluster | Predicted Core Structure(s) * | Similarity (%) |
|---|---|---|---|---|---|---|---|---|
| ptg000001l | r1c1 | 141,995 | 181,332 | 39,338 | NRPS | α-acorenol | 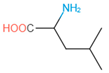 | 100 |
| r1c3 | 280,374 | 326,691 | 46,318 | T1PKS | monascorubrin | 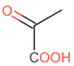 | 100 | |
| r1c10 | 2,330,446 | 2,375,712 | 45,267 | T1PKS | alternariol |  | 100 | |
| r1c15 | 6,578,008 | 6,611,156 | 33,149 | Terpene | koraiol | NA | 100 | |
| ptg000006l | r6c12 | 6,311,961 | 6,332,174 | 20,214 | Indole | sespendole | NA | 83 |
| ptg000007l | r7c3 | 2,657,144 | 2,703,633 | 46,490 | T1PKS | wortmanamide A/ wortmanamide B |  | 83 |
| ptg000008l | r8c3 | 1,389,085 | 1,431,968 | 42,884 | T1PKS | (-)-Mellein | 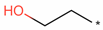 | 100 |
| Gene ID | Predicted Effector Proteins | Effector Probability |
|---|---|---|
| Dmahothocarpusptg000001lG002370.1 | Probable endo-beta-1,4-glucanase D (Precursor) | 0.691 |
| Dmahothocarpusptg000001lG005880.1 | Putative ec86 protein | 0.739 |
| Dmahothocarpusptg000001lG007160.1 | Acetylxylan esterase 2 (Precursor) | 0.787 |
| Dmahothocarpusptg000002lG000540.1 | CFEM domain-containing protein | 0.713 |
| Dmahothocarpusptg000002lG002080.1 | Protein CAP22 | 0.558 |
| Dmahothocarpusptg000002lG003240.1 | Probable pectate lyase E (Precursor) | 0.847 |
| Dmahothocarpusptg000003lG000940.1 | Putative bys1 domain protein | 0.641 |
| Dmahothocarpusptg000003lG003280.1 | Lysine-specific metallo-endopeptidase | 0.812 |
| Dmahothocarpusptg000003lG006090.1 | Putative sterigmatocystin biosynthesis peroxidase stcC | 0.681 |
| Dmahothocarpusptg000003lG006750.1 | Cryparin (Precursor) | 0.625 |
| Dmahothocarpusptg000003lG008560.1 | Putative sterigmatocystin biosynthesis peroxidase stcC | 0.573 |
| Dmahothocarpusptg000003lG008870.1 | Phosphatidylglycerol/phosphatidylinositol transfer protein | 0.787 |
| Dmahothocarpusptg000003lG011640.1 | CFEM domain | 0.647 |
| Dmahothocarpusptg000003lG016500.1 | Putative transmembrane emp24 domain-containing protein 9 protein | 0.650 |
| Dmahothocarpusptg000003lG017280.1 | Deoxyribonuclease NucA/NucB | 0.710 |
| Dmahothocarpusptg000003lG019450.1 | Pectate lyase plyB (Precursor) | 0.626 |
| Dmahothocarpusptg000004lG000540.1 | Putative carbohydrate-binding-like protein | 0.558 |
| Dmahothocarpusptg000004lG006290.1 | short chain dehydrogenase | 0.551 |
| Dmahothocarpusptg000004lG006480.1 | Pyranose dehydrogenase 3 | 0.697 |
| Dmahothocarpusptg000004lG014360.1 | Necrosis-inducing protein (NPP1) | 0.650 |
| Dmahothocarpusptg000004lG018200.1 | Probable glutamine amidotransferase SNO1 | 0.552 |
| Dmahothocarpusptg000004lG021380.1 | Pathogen effector; putative necrosis-inducing factor | 0.617 |
| Dmahothocarpusptg000004lG022680.1 | CoA binding domain | 0.646 |
| Dmahothocarpusptg000004lG024050.1 | CVNH domain | 0.886 |
| Dmahothocarpusptg000004lG033200.1 | Fungal fucose-specific lectin | 0.618 |
| Dmahothocarpusptg000005lG001070.1 | Acetylxylan esterase-like protein | 0.552 |
| Dmahothocarpusptg000005lG005760.1 | Cysteine-rich secretory protein family | 0.737 |
| Dmahothocarpusptg000005lG007250.1 | Parallel beta-helix repeat protein | 0.726 |
| Dmahothocarpusptg000005lG008360.1 | Cerato-ulmin (Precursor) | 0.699 |
| Dmahothocarpusptg000005lG012990.1 | Putative exo-beta-glucanase protein | 0.639 |
| Dmahothocarpusptg000005lG014470.1 | Pectate lyase F | 0.838 |
| Dmahothocarpusptg000006lG000360.1 | IDI-2 precursor | 0.814 |
| Dmahothocarpusptg000006lG004760.1 | Short chain dehydrogenase | 0.862 |
| Dmahothocarpusptg000006lG004870.1 | Galactan endo-beta-1,3-galactanase (Precursor) | 0.597 |
| Dmahothocarpusptg000006lG006270.1 | Aromatic peroxygenase | 0.654 |
| Dmahothocarpusptg000006lG008080.1 | Phospholipase A2 | 0.570 |
| Dmahothocarpusptg000006lG015010.1 | Pathogen effector; putative necrosis-inducing factor | 0.729 |
| Dmahothocarpusptg000006lG022730.1 | Chloroperoxidase-like protein | 0.703 |
| Dmahothocarpusptg000007lG001170.1 | chitin deacetylase | 0.586 |
| Dmahothocarpusptg000007lG001450.1 | Necrosis-inducing protein (NPP1) | 0.652 |
| Dmahothocarpusptg000007lG002990.1 | Putative endo-beta-1,4-glucanase D | 0.713 |
| Dmahothocarpusptg000007lG003330.1 | Pectate lyase D | 0.68 |
| Dmahothocarpusptg000007lG006760.1 | Ribonuclease clavin (Precursor) | 0.901 |
| Dmahothocarpusptg000008lG001670.1 | Hydrophobic surface binding protein A | 0.840 |
| Dmahothocarpusptg000008lG012160.1 | Putative barwin-like endoglucanase protein | 0.682 |
| Dmahothocarpusptg000008lG014260.1 | Putative chitin binding protein | 0.657 |
| Species | Strain | Host | Next-Generation Sequencing | BUSCO Completeness (%) | Genome Size (Mb) | GC Content (%) | Predicted Genes | CAZymes | References |
|---|---|---|---|---|---|---|---|---|---|
| D. mahothocarpus | GZU-Y2 | Camellia oleifera | Pacbio Sequel II | 97.93 | 58.97 | 50.65 | 15,918 | 1058 | This study |
| D. eres | CBS 160.32 | Blueberry | Illumina HiSeq | 98.40 | 60.80 | 47.60 | 16,499 | 859 | [55] |
| D. aspalathi | MS-SSC91 | Soybean | Illumina HiSeq 2000 | 97.60 | 55.00 | 51.00 | 14,962 | ND | [56] |
| D. citri | Q7 | Citrus | Illumina HiSeq | 98.50 | 63.61 | 47.48 | 15,422 | 1624 | [57] |
| D. citri | ZJUD14 | Citrus | Illumina HiSeq | 98.60 | 52.06 | 52.76 | 14,991 | 1581 | [57] |
| D. citri | NFHF-8-4 | Citrus | Illumina HiSeq | 97.30 | 57.00 | 46.72 | 15,921 | ND | [58] |
| D. capsici | GY-Z16 | Walnut | PacBio Sequel | 98.40 | 57.60 | 51.30 | 14,425 | 843 | [59] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, X.; Zhang, Y.; Yang, J.; Chen, Y. A Genomic Sequence Resource of Diaporthe mahothocarpus GZU-Y2 Causing Leaf Spot Blight in Camellia oleifera. J. Fungi 2024, 10, 630. https://doi.org/10.3390/jof10090630
Shi X, Zhang Y, Yang J, Chen Y. A Genomic Sequence Resource of Diaporthe mahothocarpus GZU-Y2 Causing Leaf Spot Blight in Camellia oleifera. Journal of Fungi. 2024; 10(9):630. https://doi.org/10.3390/jof10090630
Chicago/Turabian StyleShi, Xulong, Yu Zhang, Jing Yang, and Yunze Chen. 2024. "A Genomic Sequence Resource of Diaporthe mahothocarpus GZU-Y2 Causing Leaf Spot Blight in Camellia oleifera" Journal of Fungi 10, no. 9: 630. https://doi.org/10.3390/jof10090630
APA StyleShi, X., Zhang, Y., Yang, J., & Chen, Y. (2024). A Genomic Sequence Resource of Diaporthe mahothocarpus GZU-Y2 Causing Leaf Spot Blight in Camellia oleifera. Journal of Fungi, 10(9), 630. https://doi.org/10.3390/jof10090630