
Direct and Indirect Protein Interactions Link FUS Aggregation to Histone Post-Translational Modification Dysregulation and Growth Suppression in an ALS/FTD Yeast Model
 and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Yeast Strains, Media, and Plasmids
2.2. Transformation of Yeast with FLAG-Tagged Histone-Modifying Enzymes via PCR Targeting
2.3. Protein Overexpression
2.4. Serial Dilution Growth Assays
2.5. Western Blotting
2.6. Co-Immunoprecipitation
2.7. Tandem Mass Spectrometry
2.8. Immunocytochemistry
2.9. Statistical Analysis
3. Results and Discussion
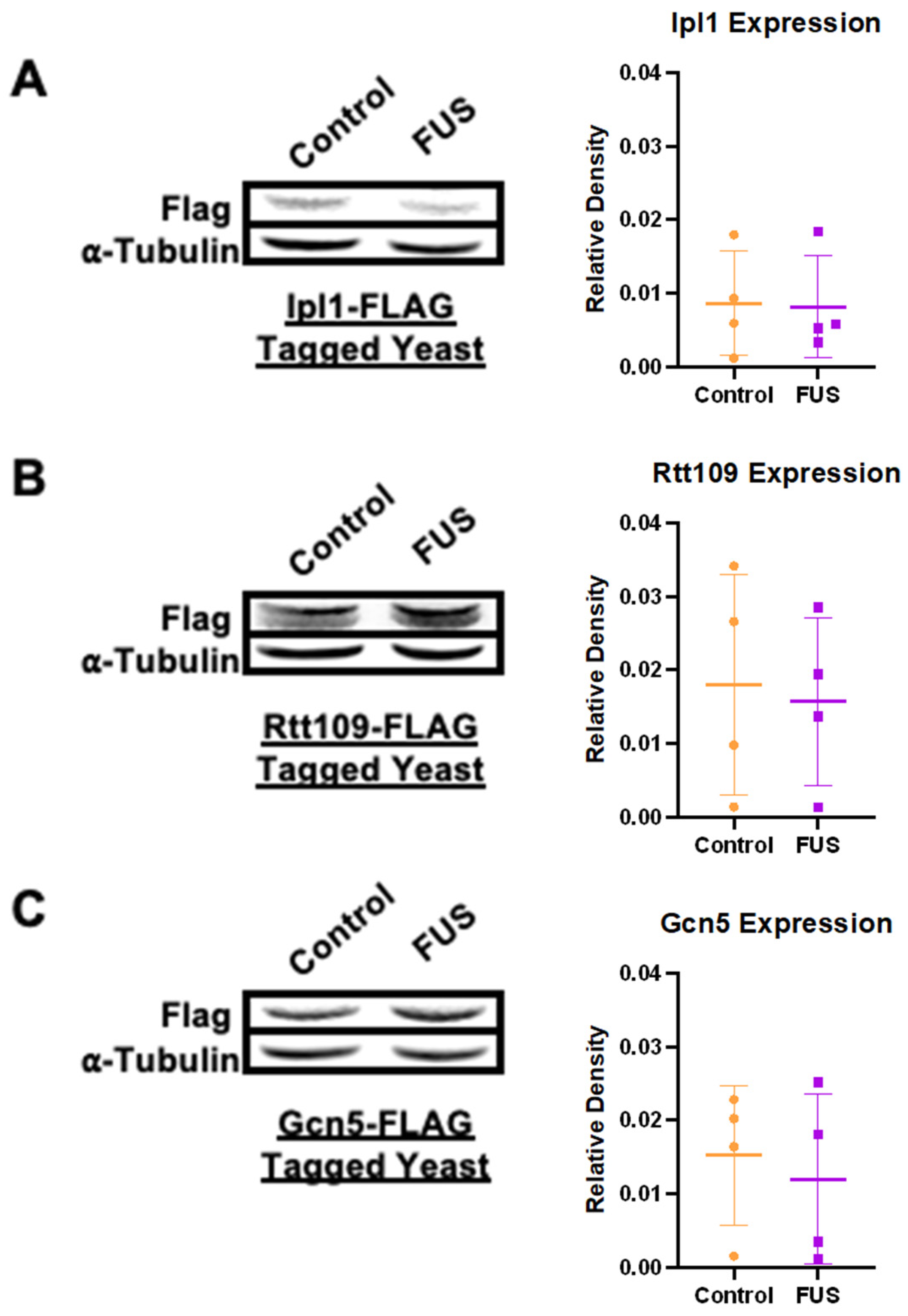
3.1. Levels of Histone-Modifying Enzymes Are Not Decreased in Yeast Overexpressing FUS
3.2. Ipl1-FLAG Colocalizes with FUS and Is Depleted from the Nucleus
3.3. Rtt109 Is Depleted from the Nucleus in Yeast Overexpressing FUS
3.4. Gcn5 Localization Is Unchanged in Yeast Overexpressing FUS
3.5. Putative Binding Partners of FUS Are Involved in ATP Binding and rRNA Processing
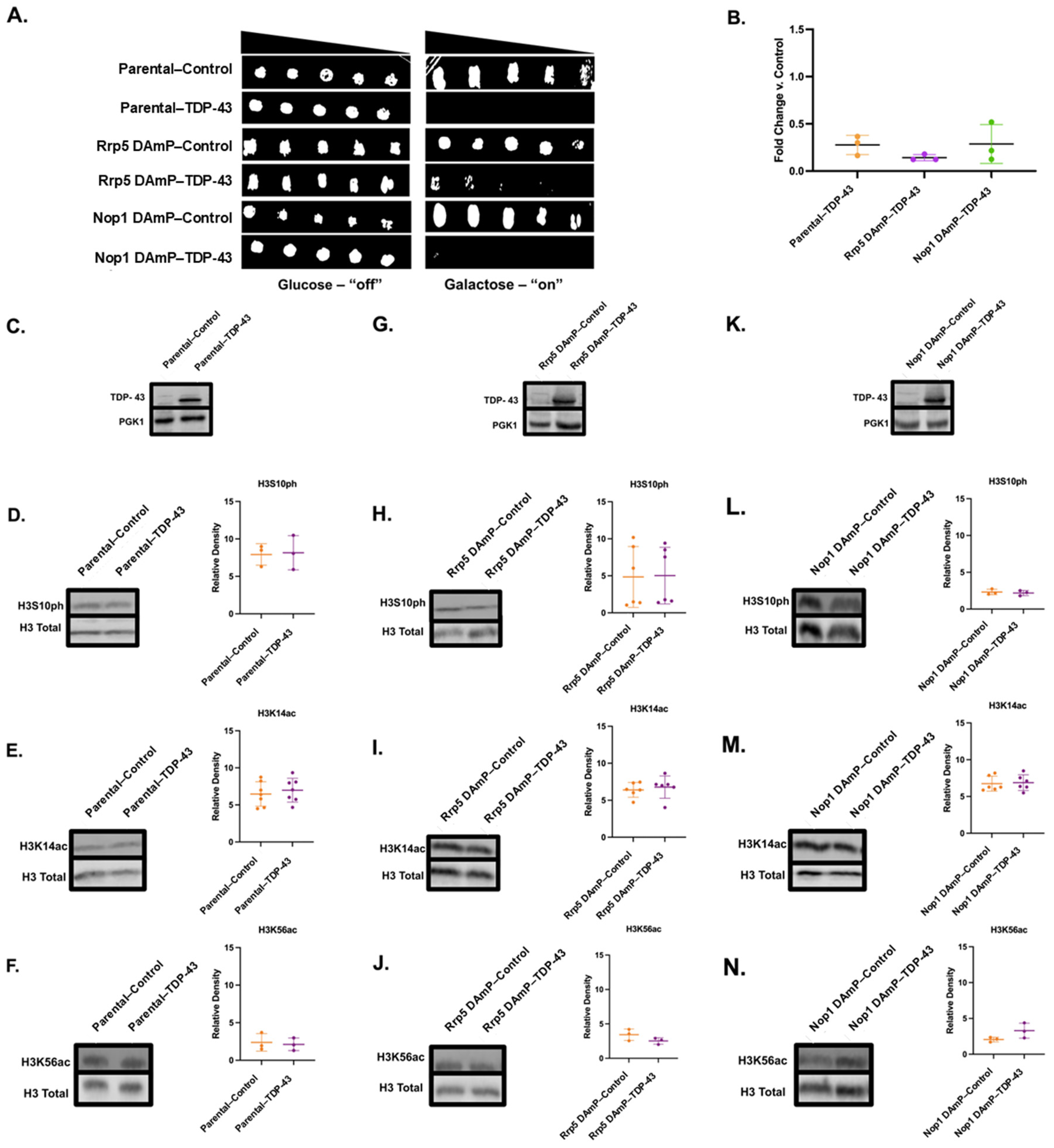
3.6. Decreased mRNA of Rrp5 Alleviates FUS-Linked Growth Suppression but Does Not Impact Histone PTM Changes
3.7. Nop1 DAmP Alleviates Growth Suppression and Restores H3S10ph and H3K14ac Levels
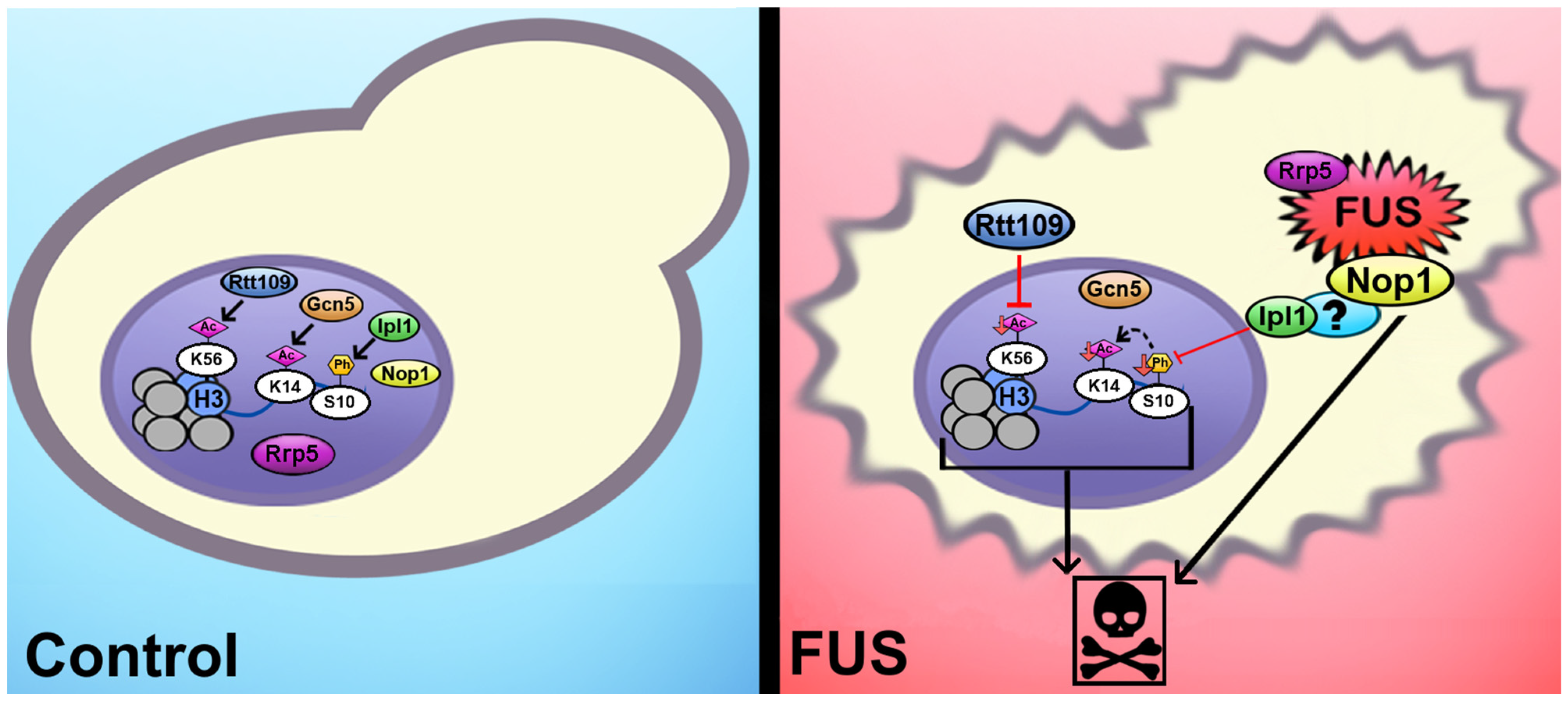
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic Lateral Sclerosis |
| Co-IP | co-immunoprecipitation |
| FTD | Frontotemporal Dementia |
| HAT | histone acetyltransferase |
| HDAC | histone deacetylase |
| HMEs | Histone Modifying Enzymes |
| ICC | immunocytochemistry |
| MS | mass spectrometry |
| PCR | polymerase chain reaction |
| PTM | post-translational modification |
References
- Pang, S.Y.-Y.; Hsu, J.S.; Teo, K.-C.; Li, Y.; Kung, M.H.W.; Cheah, K.S.E.; Chan, D.; Cheung, K.M.C.; Li, M.; Sham, P.-C.; et al. Burden of Rare Variants in ALS Genes Influences Survival in Familial and Sporadic ALS. Neurobiol. Aging 2017, 58, 238.e9–238.e15. [Google Scholar] [CrossRef]
- Nolan, M.; Talbot, K.; Ansorge, O. Pathogenesis of FUS-Associated ALS and FTD: Insights from Rodent Models. Acta Neuropathol. Commun. 2016, 4, 99. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Kapogiannis, D.; Huey, E.D.; Momeni, P. FTD and ALS: A Tale of Two Diseases. Curr. Alzheimer Res. 2011, 8, 273–294. [Google Scholar] [CrossRef]
- Debove, C.; Zeisser, P.; Salzman, P.M.; Powe, L.K.; Truffinet, P. The Rilutek (Riluzole) Global Early Access Programme: An Open-Label Safety Evaluation in the Treatment of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2001, 2, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Jorgenson, J.A.; Newhouse, B.J.; Shefner, J.M.; Agnese, W. Edaravone in the Treatment of Amyotrophic Lateral Sclerosis: Efficacy and Access to Therapy—A Roundtable Discussion. Am. J. Manag. Care 2018, 24, S175–S186. [Google Scholar] [PubMed]
- Ling, S.-C. Synaptic Paths to Neurodegeneration: The Emerging Role of TDP-43 and FUS in Synaptic Functions. Neural Plast. 2018, 2018, 8413496. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Gao, K.; Jankovic, J. The Role of FUS Gene Variants in Neurodegenerative Diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef]
- Sun, Z.; Diaz, Z.; Fang, X.; Hart, M.P.; Chesi, A.; Shorter, J.; Gitler, A.D. Molecular Determinants and Genetic Modifiers of Aggregation and Toxicity for the ALS Disease Protein FUS/TLS. PLoS Biol. 2011, 9, e1000614. [Google Scholar] [CrossRef]
- Neumann, M.; Roeber, S.; Kretzschmar, H.A.; Rademakers, R.; Baker, M.; Mackenzie, I.R.A. Abundant FUS-Immunoreactive Pathology in Neuronal Intermediate Filament Inclusion Disease. Acta Neuropathol. 2009, 118, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Doi, H.; Koyano, S.; Suzuki, Y.; Nukina, N.; Kuroiwa, Y. The RNA-Binding Protein FUS/TLS Is a Common Aggregate-Interacting Protein in Polyglutamine Diseases. Neurosci. Res. 2010, 66, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Woulfe, J.; Gray, D.A.; Mackenzie, I.R.A. FUS-Immunoreactive Intranuclear Inclusions in Neurodegenerative Disease. Brain Pathol. 2010, 20, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.A.; Lemay, N.; Ko, H.K.; Zhou, H.; Burke, C.; Kwiatkowski, T.J.; Sapp, P.; McKenna-Yasek, D.; Brown, R.H.; Hayward, L.J. Mutant FUS Proteins That Cause Amyotrophic Lateral Sclerosis Incorporate into Stress Granules. Hum. Mol. Genet. 2010, 19, 4160–4175. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The Molecular Hallmarks of Epigenetic Control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal Structure of the Nucleosome Core Particle at 2.8 Å Resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- de Ruijter, A.J.M.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B.P. Histone Deacetylases (HDACs): Characterization of the Classical HDAC Family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Huang, L. Targeting Histone Deacetylases for the Treatment of Cancer and Inflammatory Diseases. J. Cell. Physiol. 2006, 209, 611–616. [Google Scholar] [CrossRef]
- Sawicka, A.; Seiser, C. Histone H3 Phosphorylation—A Versatile Chromatin Modification for Different Occasions. Biochimie 2012, 94, 2193–2201. [Google Scholar] [CrossRef]
- Wilson, J.R.; Jing, C.; Walker, P.A.; Martin, S.R.; Howell, S.A.; Blackburn, G.M.; Gamblin, S.J.; Xiao, B. Crystal Structure and Functional Analysis of the Histone Methyltransferase SET7/9. Cell 2002, 111, 105–115. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The Language of Covalent Histone Modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Latham, J.A.; Dent, S.Y.R. Cross-Regulation of Histone Modifications. Nat. Struct. Mol. Biol. 2007, 14, 1017–1024. [Google Scholar] [CrossRef]
- Kim, S.H.; Shanware, N.P.; Bowler, M.J.; Tibbetts, R.S. Amyotrophic Lateral Sclerosis-Associated Proteins TDP-43 and FUS/TLS Function in a Common Biochemical Complex to Co-Regulate HDAC6 mRNA. J. Biol. Chem. 2010, 285, 34097–34105. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.-E.; Ko, C.-P. Treatment with Trichostatin A Initiated after Disease Onset Delays Disease Progression and Increases Survival in a Mouse Model of Amyotrophic Lateral Sclerosis. Exp. Neurol. 2011, 231, 147–159. [Google Scholar] [CrossRef]
- Corcoran, L.J.; Mitchison, T.J.; Liu, Q. A Novel Action of Histone Deacetylase Inhibitors in a Protein Aggresome Disease Model. Curr. Biol. 2004, 14, 488–492. [Google Scholar] [CrossRef]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 Inhibition Reverses Axonal Transport Defects in Motor Neurons Derived from FUS-ALS Patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef]
- Kuta, R.; Larochelle, N.; Fernandez, M.; Pal, A.; Minotti, S.; Tibshirani, M.; St Louis, K.; Gentil, B.J.; Nalbantoglu, J.N.; Hermann, A.; et al. Depending on the Stress, Histone Deacetylase Inhibitors Act as Heat Shock Protein Co-Inducers in Motor Neurons and Potentiate Arimoclomol, Exerting Neuroprotection through Multiple Mechanisms in ALS Models. Cell Stress Chaperones 2020, 25, 173–191. [Google Scholar] [CrossRef] [PubMed]
- Tejido, C.; Pakravan, D.; Bosch, L.V.D. Potential Therapeutic Role of HDAC Inhibitors in FUS-ALS. Front. Mol. Neurosci. 2021, 14, 686995. [Google Scholar] [CrossRef]
- Chen, K.; Bennett, S.A.; Rana, N.; Yousuf, H.; Said, M.; Taaseen, S.; Mendo, N.; Meltser, S.M.; Torrente, M.P. Neurodegenerative Disease Proteinopathies Are Connected to Distinct Histone Post-Translational Modification Landscapes. ACS Chem. Neurosci. 2018, 9, 838–848. [Google Scholar] [CrossRef]
- Bennett, S.A.; Cobos, S.N.; Mirzakandova, M.; Fallah, M.; Son, E.; Angelakakis, G.; Rana, N.; Hugais, M.; Torrente, M.P. Trichostatin A Relieves Growth Suppression and Restores Histone Acetylation at Specific Sites in a FUS ALS/FTD Yeast Model. Biochemistry 2021, 60, 3671–3675. [Google Scholar] [CrossRef] [PubMed]
- Rosaleny, L.E.; Ruiz-García, A.B.; García-Martínez, J.; Pérez-Ortín, J.E.; Tordera, V. The Sas3p and Gcn5p Histone Acetyltransferases Are Recruited to Similar Genes. Genome Biol. 2007, 8, R119. [Google Scholar] [CrossRef]
- Fillingham, J.; Recht, J.; Silva, A.C.; Suter, B.; Emili, A.; Stagljar, I.; Krogan, N.J.; Allis, C.D.; Keogh, M.-C.; Greenblatt, J.F. Chaperone Control of the Activity and Specificity of the Histone H3 Acetyltransferase Rtt109. Mol. Cell. Biol. 2008, 28, 4342–4353. [Google Scholar] [CrossRef] [PubMed]
- Cote, J.M.; Kuo, Y.-M.; Henry, R.A.; Scherman, H.; Krzizike, D.D.; Andrews, A.J. Two Factor Authentication: Asf1 Mediates Crosstalk between H3 K14 and K56 Acetylation. Nucleic Acids Res. 2019, 47, 7380–7391. [Google Scholar] [CrossRef]
- Schimmang, T.; Tollervey, D.; Kern, H.; Frank, R.; Hurt, E.C. A Yeast Nucleolar Protein Related to Mammalian Fibrillarin Is Associated with Small Nucleolar RNA and Is Essential for Viability. EMBO J. 1989, 8, 4015–4024. [Google Scholar] [CrossRef]
- Venema, J.; Tollervey, D. RRP5 Is Required for Formation of Both 18S and 5.8S rRNA in Yeast. EMBO J. 1996, 15, 5701–5714. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Lindquist, S.L. HSP104 Required for Induced Thermotolerance. Science 1990, 248, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Winzeler, E.A.; Shoemaker, D.D.; Astromoff, A.; Liang, H.; Anderson, K.; Andre, B.; Bangham, R.; Benito, R.; Boeke, J.D.; Bussey, H.; et al. Functional Characterization of the S. Cerevisiae Genome by Gene Deletion and Parallel Analysis. Science 1999, 285, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Jackrel, M.E.; Shorter, J. Potentiated Hsp104 Variants Suppress Toxicity of Diverse Neurodegenerative Disease-Linked Proteins. Dis. Model. Mech. 2014, 7, 1175–1184. [Google Scholar] [CrossRef]
- Bennett, S.A.; Cobos, S.N.; Meykler, M.; Fallah, M.; Rana, N.; Chen, K.; Torrente, M.P. Characterizing Histone Post-Translational Modification Alterations in Yeast Neurodegenerative Proteinopathy Models. J. Vis. Exp. 2019, 59104. [Google Scholar] [CrossRef]
- Gardner, J.M.; Jaspersen, S.L. Manipulating the Yeast Genome: Deletion, Mutation, and Tagging by PCR. In Yeast Genetics; Smith, J.S., Burke, D.J., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2014; Volume 1205, pp. 45–78. ISBN 978-1-4939-1362-6. [Google Scholar]
- Amberg, D.C.; Burke, D.J.; Strathern, J.N. Yeast Immunofluorescence. CSH Protoc. 2006, 2006, pdb.prot4167. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Kelley, J.B.; Paschal, B.M. Fluorescence-Based Quantification of Nucleocytoplasmic Transport. Methods 2019, 157, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Motulsky, H.J.; Brown, R.E. Detecting Outliers When Fitting Data with Nonlinear Regression—A New Method Based on Robust Nonlinear Regression and the False Discovery Rate. BMC Bioinform. 2006, 7, 123. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Use R! Springer: New York, NY, USA, 2009; ISBN 978-0-387-98141-3. [Google Scholar]
- Bennett, S.A.; Cobos, S.N.; Son, E.; Segal, R.; Mathew, S.; Yousuf, H.; Torrente, M.P. Impaired RNA Binding Does Not Prevent Histone Modification Changes in a FUS ALS/FTD Yeast Model. MicroPublication Biol. 2023, 2023. [Google Scholar] [CrossRef]
- Buvelot, S.; Tatsutani, S.Y.; Vermaak, D.; Biggins, S. The Budding Yeast Ipl1/Aurora Protein Kinase Regulates Mitotic Spindle Disassembly. J. Cell Biol. 2003, 160, 329–339. [Google Scholar] [CrossRef]
- Shlevkov, E.; Basu, H.; Bray, M.-A.; Sun, Z.; Wei, W.; Apaydin, K.; Karhohs, K.; Chen, P.-F.; Smith, J.L.M.; Wiskow, O.; et al. A High-Content Screen Identifies TPP1 and Aurora B as Regulators of Axonal Mitochondrial Transport. Cell Rep. 2019, 28, 3224–3237.e5. [Google Scholar] [CrossRef] [PubMed]
- Sterner, D.E.; Berger, S.L. Acetylation of Histones and Transcription-Related Factors. Microbiol. Mol. Biol. Rev. MMBR 2000, 64, 435–459. [Google Scholar] [CrossRef]
- Rouaux, C.; Jokic, N.; Mbebi, C.; Boutillier, S.; Loeffler, J.-P.; Boutillier, A.-L. Critical Loss of CBP/P300 Histone Acetylase Activity by Caspase-6 during Neurodegeneration. EMBO J. 2003, 22, 6537–6549. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Holbert, M.A.; Wurtele, H.; Meeth, K.; Rocha, W.; Gharib, M.; Jiang, E.; Thibault, P.; Verreault, A.; Cole, P.A.; et al. Fungal Rtt109 Histone Acetyltransferase Is an Unexpected Structural Homolog of Metazoan P300/CBP. Nat. Struct. Mol. Biol. 2008, 15, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Stejskal, S.; Stepka, K.; Tesarova, L.; Stejskal, K.; Matejkova, M.; Simara, P.; Zdrahal, Z.; Koutna, I. Cell Cycle-Dependent Changes in H3K56ac in Human Cells. Cell Cycle Georget. Tex 2015, 14, 3851–3863. [Google Scholar] [CrossRef]
- Tibshirani, M.; Tradewell, M.L.; Mattina, K.R.; Minotti, S.; Yang, W.; Zhou, H.; Strong, M.J.; Hayward, L.J.; Durham, H.D. Cytoplasmic Sequestration of FUS/TLS Associated with ALS Alters Histone Marks through Loss of Nuclear Protein Arginine Methyltransferase 1. Hum. Mol. Genet. 2015, 24, 773–786. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Kumar, M.S.; Ramesh, N.; Anderson, E.N.; Nguyen, A.T.; Kim, B.; Cheung, S.; McDonough, J.A.; Skarnes, W.C.; Lopez-Gonzalez, R.; et al. Interactions between ALS-Linked FUS and Nucleoporins Are Associated with Defects in the Nucleocytoplasmic Transport Pathway. Nat. Neurosci. 2021, 24, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Taylor, J.P. Lost in Transportation: Nucleocytoplasmic Transport Defects in ALS and Other Neurodegenerative Diseases. Neuron 2017, 96, 285–297. [Google Scholar] [CrossRef]
- Chen, C.-C.; Carson, J.J.; Feser, J.; Tamburini, B.; Zabaronick, S.; Linger, J.; Tyler, J.K. Acetylated Lysine 56 on Histone H3 Drives Chromatin Assembly after Repair and Signals for the Completion of Repair. Cell 2008, 134, 231–243. [Google Scholar] [CrossRef]
- Karányi, Z.; Hornyák, L.; Székvölgyi, L. Histone H3 Lysine 56 Acetylation Is Required for Formation of Normal Levels of Meiotic DNA Breaks in S. Cerevisiae. Front. Cell Dev. Biol. 2020, 7, 364. [Google Scholar] [CrossRef]
- Chen, H.; Fan, M.; Pfeffer, L.M.; Laribee, R.N. The Histone H3 Lysine 56 Acetylation Pathway Is Regulated by Target of Rapamycin (TOR) Signaling and Functions Directly in Ribosomal RNA Biogenesis. Nucleic Acids Res. 2012, 40, 6534–6546. [Google Scholar] [CrossRef]
- Workman, J.J.; Chen, H.; Laribee, R.N. Saccharomyces Cerevisiae TORC1 Controls Histone Acetylation by Signaling Through the Sit4/PP6 Phosphatase to Regulate Sirtuin Deacetylase Nuclear Accumulation. Genetics 2016, 203, 1733–1746. [Google Scholar] [CrossRef] [PubMed]
- Lo, W.S.; Trievel, R.C.; Rojas, J.R.; Duggan, L.; Hsu, J.Y.; Allis, C.D.; Marmorstein, R.; Berger, S.L. Phosphorylation of Serine 10 in Histone H3 Is Functionally Linked in Vitro and in Vivo to Gcn5-Mediated Acetylation at Lysine 14. Mol. Cell 2000, 5, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Rundlett, S.E.; Carmen, A.A.; Kobayashi, R.; Bavykin, S.; Turner, B.M.; Grunstein, M. HDA1 and RPD3 Are Members of Distinct Yeast Histone Deacetylase Complexes That Regulate Silencing and Transcription. Proc. Natl. Acad. Sci. USA 1996, 93, 14503–14508. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.M. A Ranking-Based Scoring Function for Peptide-Spectrum Matches. J. Proteome Res. 2009, 8, 2241–2252. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics Enrichment Tools: Paths toward the Comprehensive Functional Analysis of Large Gene Lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Day-Richter, J.; Harris, M.A.; Haendel, M.; Gene Ontology OBO-Edit Working Group; Lewis, S. OBO-Edit—An Ontology Editor for Biologists. Bioinformatics 2007, 23, 2198–2200. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. Toward Understanding the Origin and Evolution of Cellular Organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating Viruses and Cellular Organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Kamelgarn, M.; Chen, J.; Kuang, L.; Arenas, A.; Zhai, J.; Zhu, H.; Gal, J. Proteomic Analysis of FUS Interacting Proteins Provides Insights into FUS Function and Its Role in ALS. Biochim. Biophys. Acta 2016, 1862, 2004–2014. [Google Scholar] [CrossRef] [PubMed]
- Senissar, M.; Le Saux, A.; Belgareh-Touzé, N.; Adam, C.; Banroques, J.; Tanner, N.K. The DEAD-Box Helicase Ded1 from Yeast Is an mRNP Cap-Associated Protein That Shuttles between the Cytoplasm and Nucleus. Nucleic Acids Res. 2014, 42, 10005–10022. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Ma, W.K.; Tran, E.J. The DDX5/Dbp2 Subfamily of DEAD-Box RNA Helicases. Wiley Interdiscip. Rev. RNA 2019, 10, e1519. [Google Scholar] [CrossRef]
- Sheu, Y.-J.; Kinney, J.B.; Lengronne, A.; Pasero, P.; Stillman, B. Domain within the Helicase Subunit Mcm4 Integrates Multiple Kinase Signals to Control DNA Replication Initiation and Fork Progression. Proc. Natl. Acad. Sci. USA 2014, 111, E1899–E1908. [Google Scholar] [CrossRef]
- Tauchert, M.J.; Fourmann, J.-B.; Lührmann, R.; Ficner, R. Structural Insights into the Mechanism of the DEAH-Box RNA Helicase Prp43. eLife 2017, 6, e21510. [Google Scholar] [CrossRef]
- Reinders, J.; Zahedi, R.P.; Pfanner, N.; Meisinger, C.; Sickmann, A. Toward the Complete Yeast Mitochondrial Proteome: Multidimensional Separation Techniques for Mitochondrial Proteomics. J. Proteome Res. 2006, 5, 1543–1554. [Google Scholar] [CrossRef]
- Hasin, N.; Cusack, S.A.; Ali, S.S.; Fitzpatrick, D.A.; Jones, G.W. Global Transcript and Phenotypic Analysis of Yeast Cells Expressing Ssa1, Ssa2, Ssa3 or Ssa4 as Sole Source of Cytosolic Hsp70-Ssa Chaperone Activity. BMC Genom. 2014, 15, 194. [Google Scholar] [CrossRef] [PubMed]
- Torrente, M.P.; Shorter, J. The Metazoan Protein Disaggregase and Amyloid Depolymerase System: Hsp110, Hsp70, Hsp40, and Small Heat Shock Proteins. Prion 2013, 7, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Ju, S.; Tardiff, D.F.; Han, H.; Divya, K.; Zhong, Q.; Maquat, L.E.; Bosco, D.A.; Hayward, L.J.; Brown, R.H.; Lindquist, S.; et al. A Yeast Model of FUS/TLS-Dependent Cytotoxicity. PLoS Biol. 2011, 9, e1001052. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.H.; Davidson, I.; Matthes, H.; Garnier, J.-M.; Chambon, P. Cloning, Expression, and Transcriptional Properties of the Human Enhancer Factor TEF-1. Cell 1991, 65, 551–568. [Google Scholar] [CrossRef] [PubMed]
- Carvill, G.L.; Helbig, K.L.; Myers, C.T.; Scala, M.; Huether, R.; Lewis, S.; Kruer, T.N.; Guida, B.S.; Bakhtiari, S.; Sebe, J.; et al. Damaging de Novo Missense Variants in EEF1A2 Lead to a Developmental and Degenerative Epileptic-dyskinetic Encephalopathy. Hum. Mutat. 2020, 41, 1263–1279. [Google Scholar] [CrossRef]
- Li, L.; Ng, N.K.L.; Koon, A.C.; Chan, H.Y.E. Expanded Polyalanine Tracts Function as Nuclear Export Signals and Promote Protein Mislocalization via eEF1A1 Factor. J. Biol. Chem. 2017, 292, 5784–5800. [Google Scholar] [CrossRef]
- Murthi, A.; Shaheen, H.H.; Huang, H.-Y.; Preston, M.A.; Lai, T.-P.; Phizicky, E.M.; Hopper, A.K. Regulation of tRNA Bidirectional Nuclear-Cytoplasmic Trafficking in Saccharomyces Cerevisiae. Mol. Biol. Cell 2010, 21, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Eswara, M.B.K.; Clayton, A.; Mangroo, D. Utp22p Acts in Concert with Utp8p to Channel Aminoacyl-tRNA from the Nucleolus to the Nuclear tRNA Export Receptor Los1p but Not Msn5p. Biochem. Cell Biol. 2012, 90, 731–749. [Google Scholar] [CrossRef] [PubMed]
- Dragon, F.; Gallagher, J.E.G.; Compagnone-Post, P.A.; Mitchell, B.M.; Porwancher, K.A.; Wehner, K.A.; Wormsley, S.; Settlage, R.E.; Shabanowitz, J.; Osheim, Y.; et al. A Large Nucleolar U3 Ribonucleoprotein Required for 18S Ribosomal RNA Biogenesis. Nature 2002, 417, 967–970. [Google Scholar] [CrossRef]
- Gautier, T.; Bergès, T.; Tollervey, D.; Hurt, E. Nucleolar KKE/D Repeat Proteins Nop56p and Nop58p Interact with Nop1p and Are Required for Ribosome Biogenesis. Mol. Cell. Biol. 1997, 17, 7088–7098. [Google Scholar] [CrossRef] [PubMed]
- Gawade, K.; Plewka, P.; Häfner, S.J.; Lund, A.H.; Marchand, V.; Motorin, Y.; Szczesniak, M.W.; Raczynska, K.D. FUS Regulates a Subset of snoRNA Expression and Modulates the Level of rRNA Modifications. Sci. Rep. 2023, 13, 2974. [Google Scholar] [CrossRef] [PubMed]
- Gallo, J.-M.; Jin, P.; Thornton, C.A.; Lin, H.; Robertson, J.; D’Souza, I.; Schlaepfer, W.W. The Role of RNA and RNA Processing in Neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 10372–10375. [Google Scholar] [CrossRef] [PubMed]
- Fasolo, J.; Sboner, A.; Sun, M.G.F.; Yu, H.; Chen, R.; Sharon, D.; Kim, P.M.; Gerstein, M.; Snyder, M. Diverse Protein Kinase Interactions Identified by Protein Microarrays Reveal Novel Connections between Cellular Processes. Genes Dev. 2011, 25, 767–778. [Google Scholar] [CrossRef]
- Ptacek, J.; Devgan, G.; Michaud, G.; Zhu, H.; Zhu, X.; Fasolo, J.; Guo, H.; Jona, G.; Breitkreutz, A.; Sopko, R.; et al. Global Analysis of Protein Phosphorylation in Yeast. Nature 2005, 438, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Sweet, T.; Khalili, K.; Sawaya, B.E.; Amini, S. Identification of a Novel Protein from Glial Cells Based on Its Ability to Interact with NF-κB Subunitsr. J. Cell. Biochem. 2003, 90, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Breslow, D.K.; Cameron, D.M.; Collins, S.R.; Schuldiner, M.; Stewart-Ornstein, J.; Newman, H.W.; Braun, S.; Madhani, H.D.; Krogan, N.J.; Weissman, J.S. A Comprehensive Strategy Enabling High-Resolution Functional Analysis of the Yeast Genome. Nat. Methods 2008, 5, 711–718. [Google Scholar] [CrossRef]
- Gourse, R.L.; Gaal, T.; Bartlett, M.S.; Appleman, J.A.; Ross, W. rRNA Transcription and Growth Rate–Dependent Regulation of Ribosome Synthesis in Escherichia coli. Annu. Rev. Microbiol. 1996, 50, 645–677. [Google Scholar] [CrossRef] [PubMed]
- French, S.L.; Osheim, Y.N.; Cioci, F.; Nomura, M.; Beyer, A.L. In Exponentially Growing Saccharomyces Cerevisiae Cells, rRNA Synthesis Is Determined by the Summed RNA Polymerase I Loading Rate Rather than by the Number of Active Genes. Mol. Cell. Biol. 2003, 23, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Tiku, V.; Kew, C.; Mehrotra, P.; Ganesan, R.; Robinson, N.; Antebi, A. Nucleolar Fibrillarin Is an Evolutionarily Conserved Regulator of Bacterial Pathogen Resistance. Nat. Commun. 2018, 9, 3607. [Google Scholar] [CrossRef] [PubMed]
- Tessarz, P.; Santos-Rosa, H.; Robson, S.C.; Sylvestersen, K.B.; Nelson, C.J.; Nielsen, M.L.; Kouzarides, T. Glutamine Methylation in Histone H2A Is an RNA-Polymerase-I-Dedicated Modification. Nature 2014, 505, 564–568. [Google Scholar] [CrossRef]
- Tollervey, D.; Lehtonen, H.; Carmo-Fonseca, M.; Hurt, E.C. The Small Nucleolar RNP Protein NOP1 (Fibrillarin) Is Required for Pre-rRNA Processing in Yeast. EMBO J. 1991, 10, 573–583. [Google Scholar] [CrossRef]
- Kim, E.; Kwon, I. Phase Transition of Fibrillarin LC Domain Regulates Localization and Protein Interaction of Fibrillarin. Biochem. J. 2021, 478, 799–810. [Google Scholar] [CrossRef]
- Lee, J.; Cho, H.; Kwon, I. Phase Separation of Low-Complexity Domains in Cellular Function and Disease. Exp. Mol. Med. 2022, 54, 1412–1422. [Google Scholar] [CrossRef] [PubMed]
- Mawer, J.S.P.; Massen, J.; Reichert, C.; Grabenhorst, N.; Mylonas, C.; Tessarz, P. Nhp2 Is a Reader of H2AQ105me and Part of a Network Integrating Metabolism with rRNA Synthesis. EMBO Rep. 2021, 22, e52435. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Macias, M.I.; Moore, D.A.; Green, R.L.; Gomez-Herreros, F.; Naumann, M.; Hermann, A.; Van Damme, P.; Hafezparast, M.; Caldecott, K.W. FUS (Fused in Sarcoma) Is a Component of the Cellular Response to Topoisomerase I-Induced DNA Breakage and Transcriptional Stress. Life Sci. Alliance 2019, 2, e201800222. [Google Scholar] [CrossRef]
- Belin, S.; Beghin, A.; Solano-Gonzàlez, E.; Bezin, L.; Brunet-Manquat, S.; Textoris, J.; Prats, A.-C.; Mertani, H.C.; Dumontet, C.; Diaz, J.-J. Dysregulation of Ribosome Biogenesis and Translational Capacity Is Associated with Tumor Progression of Human Breast Cancer Cells. PLoS ONE 2009, 4, e7147. [Google Scholar] [CrossRef] [PubMed]
- Cobos, S.N.; Janani, C.; Cruz, G.; Rana, N.; Son, E.; Frederic, R.; Paredes Casado, J.; Khan, M.; Bennett, S.A.; Torrente, M.P. [PRION+] States Are Associated with Specific Histone H3 Post-Translational Modification Changes. Pathogens 2022, 11, 1436. [Google Scholar] [CrossRef]
- Cobos, S.N.; Torrente, M.P. Epidrugs in Amyotrophic Lateral Sclerosis/Frontotemporal Dementia: Contextualizing a Role for Histone Kinase Inhibition in Neurodegenerative Disease. ACS Pharmacol. Transl. Sci. 2022, 5, 134–137. [Google Scholar] [CrossRef]
- Fisher, R.M.A.; Torrente, M.P. Histone Post-Translational Modification and Heterochromatin Alterations in Neurodegeneration: Revealing Novel Disease Pathways and Potential Therapeutics. Front. Mol. Neurosci. 2024, 17, 1456052. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Gao, Y.; Dominguez, A.A.; Qi, L.S. CRISPR Technologies for Precise Epigenome Editing. Nat. Cell Biol. 2021, 23, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Ueda, J.; Yamazaki, T.; Funakoshi, H. Toward the Development of Epigenome Editing-Based Therapeutics: Potentials and Challenges. Int. J. Mol. Sci. 2023, 24, 4778. [Google Scholar] [CrossRef]
- El Hassouni, B.; Sarkisjan, D.; Vos, J.C.; Giovannetti, E.; Peters, G.J. Targeting the Ribosome Biogenesis Key Molecule Fibrillarin to Avoid Chemoresistance. Curr. Med. Chem. 2019, 26, 6020–6032. [Google Scholar] [CrossRef] [PubMed]
- Samarajeewa, A.; Jacques, B.E.; Dabdoub, A. Therapeutic Potential of Wnt and Notch Signaling and Epigenetic Regulation in Mammalian Sensory Hair Cell Regeneration. Mol. Ther. 2019, 27, 904–911. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bennett, S.A.; Cobos, S.N.; Fisher, R.M.A.; Son, E.; Frederic, R.; Segal, R.; Yousuf, H.; Chan, K.; Dansu, D.K.; Torrente, M.P. Direct and Indirect Protein Interactions Link FUS Aggregation to Histone Post-Translational Modification Dysregulation and Growth Suppression in an ALS/FTD Yeast Model. J. Fungi 2025, 11, 58. https://doi.org/10.3390/jof11010058
Bennett SA, Cobos SN, Fisher RMA, Son E, Frederic R, Segal R, Yousuf H, Chan K, Dansu DK, Torrente MP. Direct and Indirect Protein Interactions Link FUS Aggregation to Histone Post-Translational Modification Dysregulation and Growth Suppression in an ALS/FTD Yeast Model. Journal of Fungi. 2025; 11(1):58. https://doi.org/10.3390/jof11010058
Chicago/Turabian StyleBennett, Seth A., Samantha N. Cobos, Raven M. A. Fisher, Elizaveta Son, Rania Frederic, Rianna Segal, Huda Yousuf, Kaitlyn Chan, David K. Dansu, and Mariana P. Torrente. 2025. "Direct and Indirect Protein Interactions Link FUS Aggregation to Histone Post-Translational Modification Dysregulation and Growth Suppression in an ALS/FTD Yeast Model" Journal of Fungi 11, no. 1: 58. https://doi.org/10.3390/jof11010058
APA StyleBennett, S. A., Cobos, S. N., Fisher, R. M. A., Son, E., Frederic, R., Segal, R., Yousuf, H., Chan, K., Dansu, D. K., & Torrente, M. P. (2025). Direct and Indirect Protein Interactions Link FUS Aggregation to Histone Post-Translational Modification Dysregulation and Growth Suppression in an ALS/FTD Yeast Model. Journal of Fungi, 11(1), 58. https://doi.org/10.3390/jof11010058